Главная страница Случайная лекция

Мы поможем в написании ваших работ!
Порталы:
БиологияВойнаГеографияИнформатикаИскусствоИсторияКультураЛингвистикаМатематикаМедицинаОхрана трудаПолитикаПравоПсихологияРелигияТехникаФизикаФилософияЭкономика

Мы поможем в написании ваших работ!
Глава II. МЕТАБОЛИЗМ ЛИПИДОВ
Метаболизм липидов - это совокупность ферментативных реакций, протекающих в организме, исходным метаболитом (объектом изменения) которых являются липиды. Метаболизм липидов можно представить следующими основными процессами:
1. Метаболизм липидов в системе пищеварения. Это совокупность процессов тонкого эмульгирования и переваривания жира (триглицеридов) пищевых продуктов, поступающих в пищеварительный тракт с пищей, до жирных кислот, моноглицеридов и глицерина и последующее всасывание тонкоэмульгированных жиров и продуктов их расщепления из полости пищеварительного тракта в кровь и лимфу.
2. Метаболизм липидов в тканях, клетках и субклеточных структурах (митохондрии, цитозоль, эндоплазматический ретикулум и др.). Это совокупность процессов катаболизма и анаболизма липидов, протекающих в клетках, тканях и субклеточных структурах организма.
1. Катаболизм жиров
С пищей в организм ежедневно поступает от 80 до 150 г липидов. Основную массу составляют жиры, наряду с глюкозой служащие главными источниками энергии. Хотя калорийность жиров значительно выше, чем углеводов (9 по сравнению с 4,7 ккал/моль), при рациональном питании жиры обеспечивают не более 30% от общего количества калорий, поступающих с пищей. Жидкие жиры (масла) содержат в своём составе полиеновые жирные кислоты, которые не синтезируются в организме; поэтому жидкие жиры должны составлять не менее одной трети жиров пищи. С липидами в организм поступают и жирорастворимые витамины A, D, Е, К.
Триацилглицеролы – очень важный источник энергии в организме животных. В клетках они откладываются в запас в виде жировых капелек, состоящих из почти чистого жира. У животных, впадающих в спячку, и у перелетных птиц триацилглицеролы служат практически единственным источником энергии. Углеводы, если их накапливается слишком много (а способность организма хранить гликоген крайне ограничена), тоже превращаются в триацилглицеролы для длительного хранения.
Около 95% всей биологически доступной энергии в молекуле триацилглицеролов заключают в себе остатки трех жирных кислот с длинной цепью, и только 5% приходится на долю остатка глицерола. Такое большое различие в выходе энергии объясняется тем, что жирные кислоты являются значительно более высоко восстановленными соединениями. Кроме того, триацилглицеролы обладают сильно выраженной неполярностью и поэтому резервируются в почти обезвоженной форме. Количество энергии, запасенной в 1 грамме почти обезвоженного жира, более чем в 6 раз превышает количество энергии, запасенного в 1 грамме гидратированного гликогена. Именно поэтому триацилглицеролы, а не гликоген, были отобраны в ходе эволюции в качестве основного источника энергии.
У человека весом в 70 кг резервы топлива распределяются следующим образом: 100000 ккал в триацилглицеролах, 25000 ккал в белках (преимущественно в мышечных), 600 ккал в гликогене, 40 ккал в глюкозе. На жиры из общего веса тела приходится 11 кг. Если бы это же количество энергии запасалось бы в виде гликогена, общий вес тела должен был быть на 55 кг больше.
У млекопитающих, у человека основным местом накопления триацилглицеролов является цитоплазма жировых клеток. Капли триацилглицерола сливаются, образуя большие глобулы, которые могут занимать большую часть клеточного объема. Жировые клетки специализированны для синтеза и хранения триацилглицеролов, а также для их мобилизации в качестве топливных молекул, способных переноситься кровью к другим тканям.
1.1. Превращения жиров в пищеварительном тракте
Жиры составляют до 90% липидов, поступающих с пищей. В пищеварительном тракте они подвергаются целому ряду последовательных процессов, среди которых основные: эмульгирование, расщепление, всасывание. Основные этапы поступления и превращения экзогенных (поступивших в организм с пищей) жиров в организме показаны на рис.7.
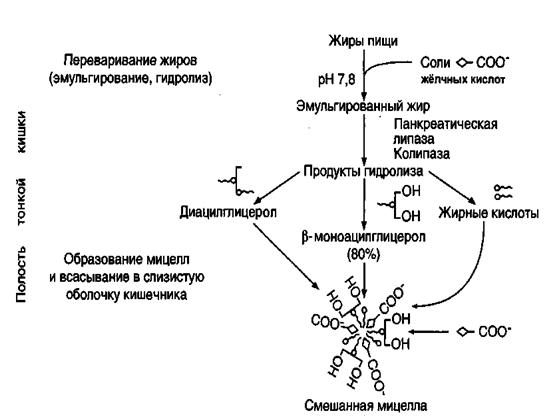
Рис.7. Основные этапы поступления в организм экзогенных жиров
(по Б.Ф.Коровину)
1.1.1. Эмульгирование жиров
Основное переваривание жиров происходит в тонком кишечнике, однако, уже в желудке небольшая часть жиров гидролизуется под действием малоактивного фермента желудочной липазы («липазы языка»). Этот фермент синтезируется железами на дорсальной поверхности языка и относительно устойчив при кислых значениях рН желудочного сока. Поэтому он действует в течение 1—2 ч на жиры пищи в желудке.
Вклад этой липазы в переваривание жиров у взрослых людей незначителен, так как:
- во-первых, в желудочном соке взрослого человека и других млекопитающих содержание липазы крайне низкое.
- во-вторых, рН желудочного сока далек от оптимума действия этого фермента (оптимальное значение рН для желудочной липазы 5,5-7,5).
- в-третьих, в желудке отсутствуют условия для эмульгирования триглицеридов, а липаза может активно действовать только на триглицериды, находящиеся в форме эмульсии.
В связи с этим у взрослых людей не эмульгированные триглицериды, составляющие основную массу пищевого жира, проходят через желудок без особых изменений.
Вместе с тем, расщепление триглицеридов в желудке играет важную роль в пищеварении у детей, особенно грудного возраста. Слизистая оболочка корня языка и примыкающей к нему области глотки ребенка грудного возраста секретирует собственную липазу в ответ на сосательные и глотательные движения. Эта липаза получила название лингвальной. Активность лингвальной липазы не успевает проявиться в ротовой полости, основным местом ее действия является желудок. Оптимум рН лингвальной липазы в пределах 4,0-4,5; он близок к величине рН желудочного сока у грудных детей.
Несмотря на то, что расщепление триглицеридов в желудке взрослого человека невелико, оно в определенной степени облегчает последующее переваривание их в кишечнике: приводит к появлению свободных жирных кислот, которые подвергаясь всасыванию в желудке, поступают в кишечник и способствуют там эмульгированию жиров, облегчая, таким образом, воздействие на нихлипазы панкреатического сока.
Основное расщепление липидов происходит в кишечнике, в первую очередь в двенадцатиперстной кишке. После того, как в нее попадает химус, происходит нейтрализация попавшей в кишечник с пищей соляной кислоты желудочного сока бикарбонатами, содержащимися в панкреатическом и кишечном соках. Выделяющиеся при разложении бикарбонатов пузырьки углекислого газа (СО2) способствуют хорошему перемешиванию пищевой кашицы с пищеварительными соками.
В этот отдел кишечника поступают также сок поджелудочной железы, содержащий ферменты расщепления жиров – панкреатические липазы, и желчь из желчного пузыря. Она содержит главным образом желчные кислоты и в небольшом количестве в ней имеются фосфолипиды и холестерол. После приёма жирной пищи желчный пузырь сокращается и желчь изливается в просвет двенадцатиперстной кишки.
Желчные кислоты выполняют в организме следующие функции: эмульгируют жиры; активируют липазу; обеспечивают всасывание высших жирных кислот, моноглицеридов и холестерина.Это обусловлено особенностями их строения, а именно тем, что онипредставляют собой поверхностно-активные вещества.
Желчные кислоты синтезируются в печени из холестерола и секретируются в желчный пузырь. Они присутствуют в желчи в конъюгированной форме, то есть в виде гликохолевой, гликодезоксихолевой, гликохенодезоксихолевой (около 2/3-4/5 всех желчных кислот) или таурохолевой, тауродезоксихолевой и таурохенодексихолевой (около 1/5-1/3 всех желчных кислот). Эти соединения иногда еще называют парными желчными кислотами, так как они состоят из двух компонентов – желчной кислоты и глицина или таурина.
Так как жиры - нерастворимые в воде соединения, то они могут подвергаться действию ферментов, растворённых в воде только на границе раздела фаз вода/жир. Поэтому действию панкреатической липазы, гидролизующей жиры, предшествует их эмульгирование.
Эмульгирование (смешивание жира с водой) происходит в тонком кишечнике под действием желчных кислот и их солей.
Считают, что только комбинация - соль желчной кислоты + ненасыщенная жирная кислота + моноглицерид - придает необходимую степень эмульгирования жира, в результате этого процесса крупные капли жира распадаются на множество мелких.
Соли желчных кислот резко уменьшают поверхностное натяжение на поверхности раздела жир/вода, благодаря чему они не только облегчают эмульгирование, но и стабилизируют уже образовавшуюся эмульсию. Эмульгированию способствует и перистальтика кишечника.
Эмульгирование приводит к увеличению площади поверхности раздела фаз жир/вода, что ускоряет гидролиз жира панкреатической липазой. В результате этого образуется очень тонкая жировая эмульсия, диаметр частиц которой не превышает 0,5 мкм. Такие эмульгированные жиры способны самостоятельно проходить через стенку кишечника и попадать в лимфатическую систему. Однако большая часть эмульгированного жира всасывается после гидролитического расщепления его панкреатическими липазами (липазами сока поджелудочной железы).
1.1.2. Расщепление жиров
Гидролиз триацилглицеролов, в результате которого освобождаются глицерин и высшие жирные кислоты, происходит постепенно под действием гидролаз эфиров глицерина — липаз (панкреатическая липаза и липаза тонкого кишечника). Активность липазы в жировой клетке регулируется гормонами.
Специфичность действия липаз определяется положением эфирных связей в триацилглицероле. Панкреатическая липаза активна к гидролизу эфирных связей в 1-м и 3-м положениях, то есть внешних сложноэфирных связей, в результате чего образуется 2-моноацилглицерол:
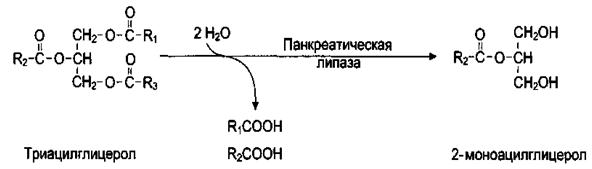
Гидролиз эфирной связи в положении 2 идет более медленно и катализируется липазой, секретируемой железами тонкого кишечника.
Образующиеся 2-моноглицеролы всасываются стенкой кишечника и либо направляются на ресинтез триглицеридов уже в кишечной стенке, либо распадаются далее под действием неспецифических эстераз. Примером может служить гидролиз β-моноглицерида (2-моноглицерола) в присутствии алиэстеразы печени:
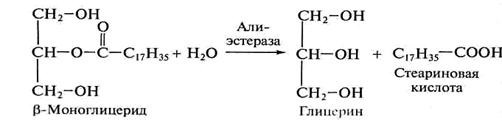
(2-моноацилглицерол)
Таким образом, схема полного гидролиза триацилглицеролов выглядит следующим образом:

Установлено, что панкреатическая липаза, как и другие пищеварительные ферменты (пепсин, трипсин, химотрипсин), поступает в верхний отдел тонкого кишечника в виде неактивной пролипазы.
Превращение пролипазы в активную липазу происходит при участии желчных кислот и еще одного белка панкреатического сока — колипазы(мол. масса около 10 кДа). Колипаза секретируется в виде проформы — проколипазы, и для ее превращения в активную колипазу требуется гидролиз специфических пептидных связей, который происходит при действии трипсина поджелудочного сока. Образовавшаяся активная колипаза образует с липазой комплекс в молярном отношении 1: 1 за счет формирования двух ионных связей лиз - глу и асп - орг. Образование такого комплекса приводит к тому, что липаза становится активной и устойчивой к действию трипсина. На скорость катализируемого липазой гидролиза жира не оказывают существенного влияния ни степень насыщенности жирных кислот, ни длина ее цепи.
Колипаза не является классическим активатором, она лишь связывает субстрат и приближает его к активному центру липазы. Колипаза своим гидрофобным доменом связывается с поверхностью мицеллы эмульгированного жира. Другая часть молекулы способствует формированию такой конформации панкреатической липазы, при которой активный центр фермента максимально приближен к своим субстратам — молекулам жиров, поэтому скорость реакции гидролиза жира резко возрастает.
Пищеварительные липазы кроме человека и млекопитающих животных обнаружены и исследованы у рыб, некоторых беспозвоночных. Однако, как правило, у большинства видов беспозвоночных и костистых рыб липолитическая активность в пищеварительных соках примерно в 1000 раз ниже, чем в панкреатическом соке млекопитающих. Не следует забывать, что жиры могут усваиваться также путем фагоцитоза и сохраняться без предварительного гидролиза до тех пор, пока не прогидролизуются внутриклеточными липазами и, таким образом, примут участие в синтезе липидов в процессах образования энергии. Выяснено, что активность липаз регулируется путем их фосфорилирования - дефосфорилирования:

Кроме жиров, с пищей поступают фосфолипиды, эфиры холестерола, однако количество этих липидов в составе пищи значительно меньше, чем жиров (~10%). Расщепление фосфолипидов происходит при участии ферментов фосфолипаз. Стериды, подвергаясь действию гидролитических ферментов типа холестераз, расщепляются в кишечнике с образованием спирта холестерола или эргостерола и соответствующей жирной кислоты. Холестеразы продуцируются поджелудочной железой и активны только в присутствии солей желчных кислот.
Таким образом, образующаяся в результате гидролиза липидов смесь содержит анионы жирных кислот, моно-, ди- и триацилглицерины, хорошо эмульгированные солями жирных кислот и мылами, глицерин, холин, этаноламин и другие полярные компоненты липидов. Исследования с мечеными триацилглицеринами показали, что около 40% жиров пищи гидролизуется полностью до глицерина и жирных кислот, 3-10% всасываются без гидролиза в форме триацилглицеринов, а остальные гидролизуются частично, главным образом до 2-моноацилглицеринов.
1.1.3. Всасывание продуктов гидролиза жиров
Образовавшийся при гидролизе жиров глицерин является водорастворимым и вместе с жирными кислотами, имеющими короткие углеродные цепи (С<10), всасывается свободно через стенку кишечника и через портальную систему кровообращения поступает в печень.
Для всасывания жирных кислот с длинной цепью (С>10), моноглицеридов необходимы желчные кислоты. Соединяясь с вышеперечисленными соединениями, желчные кислоты образуют в просвете кишечника растворимые комплексы или смешанные мицеллы - холеиновые комплексы,которые легко всасываются в эпителий кишечника.
Смешанные мицеллы построены таким образом, что гидрофобные части молекул обращены внутрь мицеллы, а гидрофильные — наружу, поэтому мицеллы хорошо растворяются в водной фазе содержимого тонкой кишки. Стабильность мицелл обеспечивается в основном солями желчных кислот. Мицеллы сближаются с каймой клеток слизистой оболочки тонкого кишечника, и липидные компоненты мицелл диффундируют через мембраны внутрь клеток. Вместе с продуктами гидролиза липидов всасываются жирорастворимые витамины A, D, Е, К и соли желчных кислот.
Желчные кислоты далее попадают через воротную вену в печень, из печени вновь секретируются в желчный пузырь и далее опять участвуют в эмульгировании жиров. Этот путь желчных кислот называют «энтерогепатическая циркуляция».Каждая молекула желчных кислот за сутки проходит 5— 8 циклов.
Так как рН в тонком кишечнике слабощелочная, желчные кислоты функционируют здесь в форме своих солей. Особую роль при этом играют такие желчные кислоты, как таурохолевая и гликохолевая. Лучше перевариваются и всасываются липиды, находящиеся в жидком состоянии, при температуре тела. Липиды, у которых точка плавления существенно выше температуры тела, плохо перевариваются и всасываются.
Фосфорная кислота, образующаяся при гидролизе фосфолипидов, всасывается в виде натриевых и калиевых солей, а азотистые основания — холин, этаноламин и серин — всасываются при участии нуклеотидов (ЦДФ-производных). Среди основных стероидов пищи только холестерин легко проникает через стенки кишечника. С такой же легкостью всасываются витамин D и некоторые стероидные гормоны.
1.1.4. Транспорт жиров из кишечника
Липиды, прежде чем поступить в лимфу, в кишечной стенке подвергаются ресинтезу, то есть превращению в триацилглицеролы. Липиды в водной среде (а значит, и в крови) нерастворимы, поэтому для транспорта холестерола и ресинтезированных липидов кровью в организме образуются комплексы липидов с белками — липопротеины (ЛП).
Все типы липопротеинов имеют сходное строение — гидрофобное ядро и гидрофильный слой на поверхности (рис.8). Гидрофильный слой образован белками, которые называют апопротеинами (аполипротеинами), и амфифильными молекулами липидов — фосфолипидами и холестеролом. Гидрофильные группы этих молекул обращены к водной фазе, а гидрофобные части — к гидрофобному ядру липопротеина, в котором находятся транспортируемые липиды.
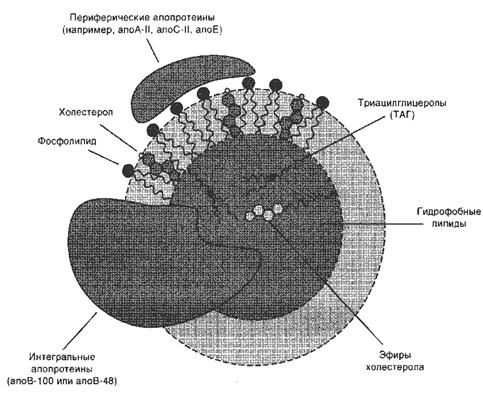
Рис.8. Липопротеины плазмы крови
Некоторые апопротеины интегральные и не могут быть отделены от липопротеина, а другие могут свободно переноситься от одного типа липопротеина к другому. Апопротеины выполняют несколько функций:
• формируют структуру липопротеинов;
• взаимодействуют с рецепторами на поверхности клеток и таким образом определяют, какими тканями будет захватываться данный тип липопротеинов;
• служат ферментами или активаторами ферментов, действующих на липопротеины.
Классификация липопротеинов основана на величине их плотности, которая в свою очередь зависит от содержания липидов. В организме синтезируются следующие типы липопротеинов: хиломикроны (ХМ), липопротеины очень низкой плотности (ЛПОНП), липопротеины промежуточной плотности (ЛППП), липопротеины низкой плотности (ЛПНП) и липопротеины высокой плотности (ЛПВП).
Липопротеины очень низкой плотности (ЛПОНП) ρ = 0,95-1г/мл, состоят на 10% из белка, 60% триацилглицеролов, 18% фосфолипидов, 15% холестерола.
Липопротеины низкой плотности (ЛПНП) ρ = 1-1,06 г/мл, состоят на 25% из белка, 10% триацилглицеролов, 22% фосфолипидов, 45% холестерола.
Липопротеины высокой плотности (ЛПВП) ρ = 1,06-1,21 г/мл, состоят на 50% из белка, 3% триацилглицеролов, 30% фосфолипидов, 18% холестерола.
Хиломикроны (ХМ) состоят на 85% из триглицеридов, поэтому вместе с липопротеинами очень низкой плотности (ЛПОНП) их относят к триглицерид-богатым липопротеинам. Кроме триглицеридов хиломикроны содержат также холестерин и эфиры холестерина. При секреции единственным белком в составе хиломикрон является одна из изоформ аполипопротеина B, который частично покрывает поверхность хиломикрона и таким образом обеспечивает стабильность частицы в процессе циркуляции.
Каждый из типов ЛПобразуется в разных тканях и транспортирует определённые липиды. Например, ХМ транспортируют экзогенные (пищевые жиры) из кишечника в ткани, поэтому триацилглицеролы составляют до 85% массы этих частиц.
ЛП хорошо растворимы в крови, некоторые из них легко проходят через стенки капилляров кровеносных сосудов и доставляют липиды к клеткам.
В плазме крови имеются три основных класса липопротеинов, причем содержание липидов в них может составлять от 50 до 90%;. Молекулы липидов и полипептидов в липопротеинах прочно соединены между собой, хотя и не образуют ковалентных связей. Липопротеины плазмы крови содержат полярные липиды и триацилглицеролы, а так же холестерол и его эфиры.
Большой размер ХМ не позволяет им проникать через стенки капилляров, поэтому из клеток кишечника они сначала попадают в лимфатическую систему и потом через главный грудной проток вливаются в кровь вместе с лимфой.
1.2. Катаболизм и энергетика глицерола
Глицерол, образованный при гидролизе триацилглицеролов, независимо от того, поступит ли он на ресинтез жиров или будет претерпевать дальнейший распад, прежде всего фосфорилируется. Донором остатка фосфорной кислоты в этой реакции служит АТФ. Процесс ускоряется соответствующей фосфотрансферазой (А):
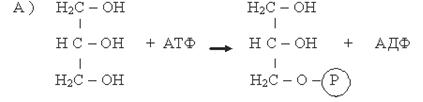
Глицерол Фосфоглицерол
Фермент: Глицеролкиназа
Образующийся фосфоглицерол, в основном, расходуется на синтез новых молекул триглицеридов, но часть его окисляется с образованием фосфодиоксиацетона (ФДА) (Б):
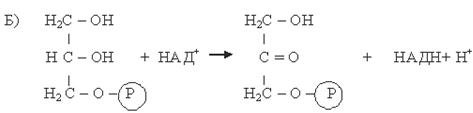
Фосфоглицерол Фосфодиоксиацетон (ФДА)
Фермент: Глицерофосфатдегидрогеназа
Фосфодиоксиацетон, в свою очередь, изомеризуется до фосфоглицеринового альдегида (ФГА) (В):
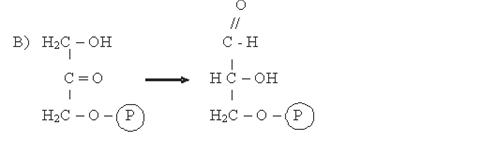
Фосфодиоксиацетон (ФДА) Фофоглицериновый альдегид (ФГА)
Фермент: Триозофосфатизомераза
ФГА служит промежуточным продуктом гликолиза и глюконеогенеза.
Таким образом, в печени, содержащей соответствующие ферменты, глицерол может превратиться в пируват (ПВК), который в аэробных условиях через цикл Кребса и окислительное фосфорилирование полностью окисляется до углекислого газа и воды. Энергия 1 моль глицерина аккумулируется (трансформируется) в 22 моль АТФ.
1.3. β - Окисление жирных кислот
Как уже указывалось, значительную часть энергии, извлекаемой в процессе окисления, животный организм получает из жирных кислот, которые расщепляются путем окисления при β-углеродном атоме.
β-Окисление жирных кислот было впервые изучено в 19004 г. Ф. Кноопом. В дальнейшем было установлено, что β-окисление осуществляется только в митохондриях. Благодаря работам Ф. Линена с сотрудниками (1954-1958 г.г.) были выяснены основные ферментативные процессы окисления жирных кислот. В честь ученых, открывших данный путь окисления жирных кислот, процесс β-окисления получил название цикла Кноопа-Линена.
β-Окисление — специфический путь катаболизма жирных кислот, при котором от карбоксильного конца жирной кислоты последовательно отделяется по 2 атома углерода в виде ацетил-КоА. Метаболический путь - β-окисление - назван так потому, что реакции окисления жирной кислоты происходят у β-углеродного атома. Реакции β-окисления и последующего окисления ацетил-КоА в ЦТК (цикле трикарбоновых кислот) служат одним из основных источников энергии для синтеза АТФ по механизму окислительного фосфорилирования. β-Окисление жирных кислот происходит только в аэробных условиях.
Все реакции многостадийного окисления ускоряются специфическими ферментами. β-окисление высших жирных кислот является универсальным биохимическим процессом, протекающим во всех живых организмах. У млекопитающих этот процесс происходит во многих тканях, в первую очередь в печени, почках и сердце. Окисление жирных кислот происходит в митохондриях. Ненасыщенные высшие жирные кислоты (олеиновая, линолевая, линоленовая и др.) предварительно восстанавливаются до предельных кислот.
Проникновению жирных кислот в митохондриальный матрикс предшествует их активация путем образования соединения с коэнзимом А (НS~КоА), содержащего макроэргическую связь. Последняя, видимо, способствует более гладкому протеканию реакций окисления образовавшегося соединения, которое называют ацилкоэнзимом А (ацил-КоА).
Взаимодействие высших жирных кислот с КоА ускоряется специфическими лигазами - ацил-КоА-синтетазами трех видов, специфичных соответственно для кислот с коротким, средним и длинным углеводородными радикалами. Они локализованы в мембранах эндоплазматической сети и в наружной мембране митохондрий. По-видимому, все ацил-КоА-синтетазы являются мультимерами; так, фермент из микросом печени имеет молекулярную массу 168 кДа и состоит из 6 идентичных субъединиц. Реакция активации жирных кислот протекает в 2 этапа:
а) сначала жирная кислота реагирует с АТФ с образаванием ациладенилата:
RCOOH + ATФ → RCO~AMФ + ФФ
б) затем идет образование активированной формы ацил-КоА:
RCO~AMФ + НS~КоА → RCO~SKoA + AMФ
Пирофосфат (ФФ) быстро гидролизуется под действием пирофосфатазы, в результате чего вся реакция оказывается необратимой: ФФ + H2O → 2Ф
Суммарное уравнение:
RCOOH + ATФ+ НS~КоА→ RCO~SKoA + AMФ + 2Ф
Жирные кислоты с короткой и средней длиной цепи (от 4 до 12 атомов углерода) могут проникать в матрикс митохондрий путём диффузии, там происходит их активация. Жирные кислоты с длинной цепью, которые преобладают в организме человека (от 12 до 20 атомов углерода), активируются ацил-КоА синтетазами, расположенными на внешней мембране митохондрий.
Внутренняя мембрана митохондрий непроницаема для длинноцепочных ацил-КоА, образовавшихся в цитоплазме. Переносчиком активированных жирных кислот служит карнитин (витамин Вт), который поступает с пищей или синтезируется из незаменимых аминокислот лизина и метионина.
В наружной мембране митохондрий находится фермент карнитинацилтрансфераза I (карнитин-палъмитоилтрансфераза I), катализи- рующий реакцию с образованием ацилкарнитина:
 H3C H3C
H3C H3C
RCO~SKoA + H3C- N+-CH2-CH-CH2-COOH ↔ H3C- N+-CH2-CH-CH2-COOH + HS~KoA
H3C H3C О
OH O-C – R
Ацил-КоА Карнитин (Вт) Ацилкарнитин Кофермент А
Этот фермент является регуляторным, он регулирует скорость поступления ацильных групп в митохондрии, а, следовательно, и скорость окисления жирных кислот.
Образовавшийся ацилкарнитин проходит через межмембранное пространство к наружной стороне внутренней мембраны и транспортируется с помощью карнитинацилкарнитинтранслоказы на внутреннюю поверхность внутренней мембраны митохондрий, где фермент карнитинацилтрансфераза II катализирует перенос ацила на внутримитохондриальный КоА, то есть обратную реакцию (рис.9).
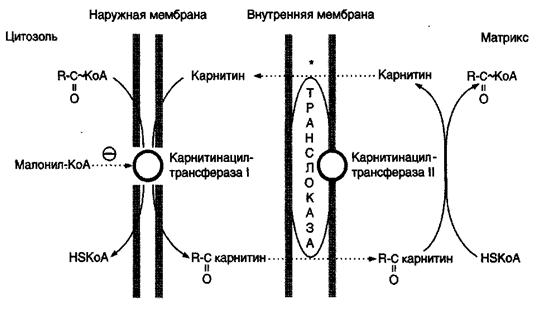
Рис.9. Перенос жирных кислот с длинным углеводородным радикалом через мембраны митохондрий
Итак, ацил-КоА становится доступным для ферментов β-окисления. Свободный карнитин возвращается на цитозольную сторону внутренней мембраны митохондрий той же транслоказой. После этого ацил-КоА включается в реакции β-окисления.
В матриксе митохондрий происходит катаболизм (распад) ацил-КоА в результате повторяющейся последовательности из четырех реакций.
1) Первой реакцией в каждом цикле является его окисление ферментом ацил-КоА-дегидрогеназой, коферментом которого является ФАД. Дегидрирование происходит между β - и α - атомами углерода, в результате чего в углеродной цепи образуется двойная связь и продуктом этой реакции является еноил-КоА:
R-CH2-CH2CO~SKoA + ФАД → R-CH=CHCO~SKoA + ФАДН2
Ацил-КоА Еноил-КоА
2) На втором этапе цикла окисления жирных кислот происходит гидратация двойной связи еноил-КоА, в результате чего образуется β-гидроксиацил-КоА. Реакция катализируется ферментом еноил-КоА-гидратазой:
OH

R-CH=CHCO~SKoA +Н2О → R-CH-CH2CO~SKoA
Еноил-КоА β- гидроксиацил-КоА
3) На третьем этапе цикла β-гидроксиацил-КоА подвергается дегидрированию (второму окислению) при участии фермента β-гидроксиацил-КоА-дегидрогеназы, коферментом которой является НАД+. Продуктом данной реакции является β-кетоацил-КоА:
OH

R-CH-CH2CO~SKoA + НАД+ → R-CОCH2CO~SKoA + НАДН + Н+
β- гидроксиацил-КоА β- кетоацил-КоА
4) Последняя реакция цикла окисления жирных кислот катализируется ацетил-КоА-ацилтрансферазой (тиолазой). На этом этапе β-кетоацил-КоА взаимодействует со свободным КоА и расщепляется с образованием, во-первых, двухуглеродного фрагмента, содержащего два концевых углеродных атома исходной жирной кислоты в виде ацетил-КоА, и, во-вторых, КоА-эфира жирной кислоты, укороченной теперь на два атома углерода. По аналогии с гидролизом эту реакцию называют тиолизом:
R-CОCH2CO~SKoA + НS~KoA → CH3CO~SKoA + R1CO~SKoA
β- кетоацил-КоА Ацетил-КоА Ацил-КоА,
укороченный на
2 углеродных атома
Укороченный ацил-КоА подвергается далее следующему циклу окисления, начинающемуся с реакции, катализируемой ацил-КоА-дегидрогеназой (окисление), затем следует реакция гидратации, реакция второго окисления, тиолазная реакция, то есть этот процесс многократно повторяется (рис.10).
β- Окисление высших жирных кислот протекает в митохондриях. В них же локализованы ферменты дыхательного цикла, ведущие передачу атомов водорода и электронов на кислород в условиях окислительного фосфорилирования АДФ, поэтому β-окисление высших жирных кислот является источником энергии для синтеза АТФ.
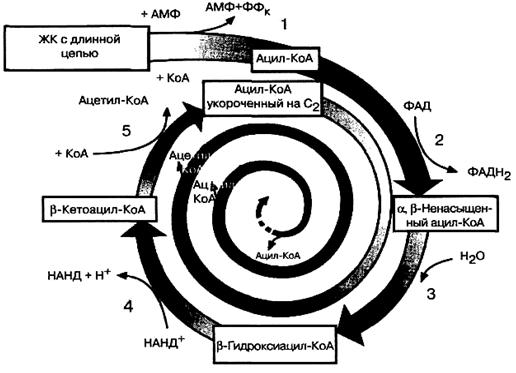
Рис.10. Окисление жирной кислоты
Окончательным продуктом β-окисления высших жирных кислот с четным числом углеродных атомов является ацетил-КоА, а с нечетным - пропионил-КоА.
Если бы ацетил-КоА накапливался в организме, то запасы HS~KoA скоро исчерпались бы, и окисление высших жирных кислот остановилось. Но этого не происходит, так как КоА быстро освобождается из состава ацетил-КоА. К этому приводит ряд процессов: ацетил-КоА включается в цикл трикарбоновых и дикарбоновых кислот или весьма близкий к нему глиоксилевый цикл, или ацетил-КоА используется для синтеза стеролов и соединений, содержащих изопреноидные группировки и т.п.
Пропионил-КоА, являющийся конечным продуктом β-окисления высших жирных кислот с нечетным числом углеродных атомов, превращается в сукцинил-КоА, который утилизируется через цикл трикарбоновых и дикарбоновых кислот.
Около половины жирных кислот в организме человека ненасыщенные.
β-Окисление этих кислот идёт обычным путём до тех пор, пока двойная связь не окажется между третьим и четвёртым атомами углерода. Затем фермент еноил-КоА-изомераза перемещает двойную связь из положения 3—4 в положение 2-3 и изменяет цис-конформацию двойной связи на транс-, которая требуется для β-окисления. В этом цикле β-окисления первая реакция дегидрирования не происходит, так как двойная связь в радикале жирной кислоты уже имеется. Далее циклы β-окисления продолжаются, не отличаясь от обычного пути. Основные пути метаболизма жирных кислот демонстрирует ри.11.
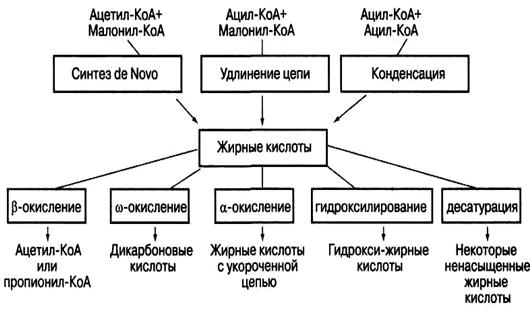
Рис.11.Основные пути метаболизма жирных кислот
Недавно было обнаружено, что помимо β-окисления – основного пути катаболизма жирных кислот, в тканях мозга происходит α-окисление жирных кислот с числом атомов углерода (С13-С18), то есть последовательное отщепление одноуглеродных фрагментов от карбоксильного конца молекулы.
Этот тип окисления наиболее характерен для растительных тканей, но может происходить и в некоторых тканях животных. α-Окисление имеет циклический характер, причем цикл состоит из двух реакций.
Первая реакция заключается в окислении жирной кислоты пероксидом водорода в соответствующий альдегид и СО2 с участием специфической пероксидазы:

В результате этой реакции углеводородная цепь укорачивается на один атом углерода.
Суть второй реакции заключается в гидратации и окслении образовавшегося альдегида в соответствующую карбоновую кислоту под действием альдегиддегидрогеназы, содержащей окисленную форму кофермента НАД:

Затем цикл α-окисления повторяется снова. В сравнении с β-окислением этот тип окисления энергетически менее выгоден.
ω-Окисление жирных кислот. В печени животных и у некоторых микроорганизмов существует ферментная система, обеспечивающая ω-окисление жирных кислот, то есть окисление по концевой СН3-группе, обозначаемой буквой ω. Сначала под действием монооксигеназы происходят гидроксилирование с образованием ω-оксикислоты:
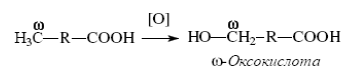
Затем ω-оксикислота окисляется в ω-дикарбоновую кислоту под действием соответствующей дегидрогеназы:

Полученная таким образом ω-дикарбоновая кислота укорачивается с любого конца с помощью реакций β-окисления.
1.4. Энергетика процессов катаболизма жиров
В каждом цикле реакций ацил-КоА укорачивается на два углеродных атома и образуется по одной молекуле ФАДН2, НАДН+Н+ и ацетил-КоА. Жирная кислота, содержащая n-число углеродных атомов претерпевает (n/2 -1) циклов. При окислении одной молекулы НАДН+Н+ через дыхательную цепь образуется 3 АТФ, а при окислении одной молекулы ФАДН2 – 2 АТФ. Окисление ацетил-КоА (n/2) в цикле Кребса и дыхательной цепи дает 12 АТФ.
Отсюда энергетический баланс окисления высшей жирной кислоты рассчитывается по следующей формуле:
[(n/2-1) x 5 + n/2 x 12 – 1],
где n – число углеродных атомов в составе высшей жирной кислоты;
n/2 – число молекул ацетил-КоА, образовавшихся при β-окислении высшей жирной кислоты;
(n/2 -1) – число циклов β-окисления, которое претерпевает данная высшая жирная кислота;
5 – число молекул АТФ, синтезирующихся за 1 цикл β-окисления высшей жирной кислоты;
12 - число молекул АТФ, синтезирующихся из 1-ой молекулы ацетил-КоА в цикле Кребса и окислительном фосфорилировании;
(-1) – молекула АТФ, которая затрачивается на активацию высшей жирной кислоты
Так, например, при β-окислении 1 моль пальмитиновой кислоты синтезируется 130 молекул АТФ, что видно по результатам вычисления по данной формуле: (16/2-1) x 5 + 16/2 x 12 – 1 = 130).
Для расчета энергетического баланса окисления триацилглицерола (простого жира) можно вывести формулу:
[(n/2 - 1) x 5 + n/2 x 12 – 1] x 3 +22,
где n – число углеродных атомов в составе высшей жирной кислоты;
n/2 – число молекул ацетил-КоА, образовавшихся при β-окислении высшей жирной кислоты;
(n/2 -1) – число циклов β-окисления, которое претерпевает данная высшая жирная кислота;
5 – число молекул АТФ, синтезирующихся за 1 цикл β-окисления высшей жирной кислоты;
12 - число молекул АТФ, синтезирующихся из 1-ой молекулы ацетил-КоА в цикле Кребса и окислительном фосфорилировании;
(-1) – молекула АТФ, которая затрачивается на активацию высшей жирной кислоты;
3 – число ацильных радикалов в составе любого жира;
22 – число молекул АТФ, синтезирующихся при катаболизме 1 моль глицерола.
Например, энергетический баланс трипальмитилглицерола составляет 412 молекул АТФ, что видно из результатов вычисления по данной формуле: (16/2-1) x 5 + 16/2 x 12 – 1) x 3 +22 = 412).
Эффективность накопления энергии в результате окисления жирных кислот при стандартных условиях составляет около 40%, что близко к этой величине для гликолиза, ЦТК, окислительного фосфорилирования.
2. Синтез и ресинтез жиров
Для обмена жиров характерно широкое использование продуктов их распада для ресинтеза. В связи с этим значительная часть β-моноглицеридов (2-моноацилглицеролов), глицерина и свободных высших жирных кислот, освобождающихся при гидролизе триглицеридов, используется для ресинтеза триглицеридов, но несколько иного состава и строения, характерного для того или иного организма (если для этого использовать пищевые жиры) или органа (если идет перестройка жиров в пределах организма).
Продукты распада жиров всасываются через стенки кишок в лимфу и поступают в кровь. В стенке кишечника происходит ресинтез нейтрального жира; из чужеродного жира образуется жир, свойственный данному виду организма. В известной мере это обеспечивается тем, что в синтезе триацилглицеролов, а также фосфолипидов в кишечной стенке принимают участие наряду с экзогенными и эндогенные жирные кислоты.
Однако способности к осуществлению в стенке кишечника синтеза жира, специфичного для данного вида животного все же ограничена. Основная масса его откладывается в жировых депо: подкожной клетчатке, сальнике, брыжжейке и жировых прослойках различных органов. Этот резервный жир расходуется при недостатке в пище, и в первую очередь при истощении углеводистых ресурсов. Более длительная способность переносить голодание обусловлена наличием жировых депо.
Липиды, входящие в состав протоплазмы клеток других органов и тканей отличаются высокой специфичностью, их состав и свойства мало зависят от пищевых жиров.
Как правило, отличие синтеза и ресинтеза жиров сводится к тому, что в ресинтезе участвуют экзогенные высшие жирные кислоты, а в синтезе эндогенные высшие жирные кислоты. Кроме того, ресинтез может протекать из продуктов неполного или полного гидролиза экзогенных жиров. Основные этапы этих процессов могут быть сходны.
2.1.Синтез нейтральных жиров
При синтезе нейтральных жиров отправными веществами являются ацил-КоА и фосфоглицерол. Фосфоглицерол образуется при фосфорилировании глицерина или при восстановлении фосфодиоксиацетона (ФДА). Ацил-КоА возникает в процессе синтеза высших жирных кислот, а также путем активирования высших жирных кислот при β-окислении.
Процесс синтеза жиров осуществляется посредством реакций трансацилирования и складывается из нескольких этапов:
1. Образование фосфоглицерола;
2. Активация высших жирных кислот, синтезированных из ацетил-КоА (эндогенного происхождения);
3. Трансферазная реакция - реакция трансацилирования – идущая в две стадии (перенос 2-х ацильных радикалов высших жирных кислот с ацил-КоА на глицерофосфат) с образованием фосфатидной кислоты;
4. Гидролиз фосфатидной кислоты с образованием диацилглицерола;
5. Трансферазная реакция - реакция трансацилирования - (перенос еще одного ацильного радикала высшей жирной кислоты с ацил-КоА на третий углеродный атом диацилглицерола) с образованием триацилглицерола.
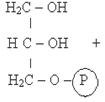
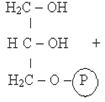
1)Фософоглицерол образуется или за счет фосфорилирования глицерина в киназной реакции (А) или при восстановлении промежуточного продукта гликолиза фосфодиоксиацетона (ФДА) (Б).
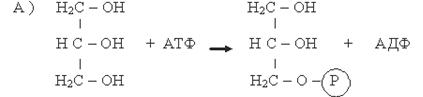
Глицерол Фосфоглицерол
Фермент: Глицеролкиназа
 Б) H2C-OH
Б) H2C-OH
HC-OН + НАДН +Н+ → НАД+
H2C-OH
Фосфодиоксиацетон (ФДА) Фосфоглицерол
Фермент: Глицерофосфатдегидрогеназа
Прямое фосфорилирование глицерола (А) характерно для почек животных, стенок кишечника, а также для микроорганизмов. В жировой ткани, мышцах идет образование глицерол-3-фосфата через гликолиз (Б). В клетках печени возможно образование глицерофосфата двумя путями.
2)Жирные кислоты, синтезированные путем новообразования из ацетил-КоА, активируются под действием фермента ацил-КоА-синтетазы и превращаются в ацил-КоА (подробнее см. β-окисление жирных кислот):
R-COOH + HS~KoA + АТФ → R-CO ~ SKoA + АМФ + 2 H3PO4
3)На третьем этапе идет трансацилирование двух свободных гидроксильных групп глицерофосфата двумя молекулами КоА-производных жирных кислот с образованием фосфатидной кислоты при участии фермента глицерофосфатацилтрансфераза:
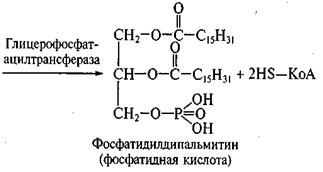
2С15Н31СО~ SKoA
Глицерофосфат Пальмитил-КоА
4)При участии фермента фосфатазы фосфатидная кислота гидролизуется с образованием диглицерида и фосфорной кислоты:

Фосфатидная кислота Дипальмитилглицерол
5)Диацилглицерол, взаимодействуя с третьей молекулой КоА-производного жирной кислоты (R-CO~SKoA) в результате трансацилазной реакции под действием фермента диглицеридацилтрансфераза (трансацилаза) превращается в триацилглицерол:

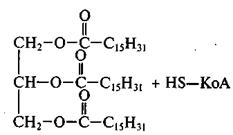
Дипальмитинглицерол Трипальмитинглицерол
Формирование каждой эфирной связи триацилглицеролов требует значительного количества свободной энергии. При активизации жирной кислоты используется энергия двух высокоэнергетических фосфатных связей благодаря пирофосфатному расщеплению АТФ и последующему гидролизу пирофосфата.
В норме у взрослых людей и у животных биосинтез и окисление жиров протекает одновременно, и для этих процессов устанавливается определенное стационарное состояние. В связи с этим количество жира в организме сохраняется в течение сравнительно длительного времени на относительно постоянном уровне, хотя, конечно, при изменении калорийности пищевого рациона могут возникать незначительные временные отклонения. Однако в тех случаях, когда углеводы, жиры или белки употребляются в количествах, превосходящих энергетические потребности организма, излишки калорий запасаются в виде триацилглицеролов.
2.2. Механизм ресинтеза жиров
Ресинтез жиров может протекать из продуктов неполного (2-моноацилглицеролов) и полного (экзогенные жирные кислоты, глицерол) гидролиза пищевых жиров.
Основной механизм ресинтеза триацилглицеролов в клетках стенки кишечника в общих чертах сводится к следующему:
- первоначально из экзогенных жирных кислот образуется их активная форма ацил-КоА при участии фермента ацил-КоА-синтетаза (А);
- после чего происходит ацилирование 2-моноацилглицеролов с образованием диацилглицеролов (Б) под действием ферментов ацил-КоА-трансфераз;
- затем под действием этих же ферментов образование триацилглицеролов (В).
А) С17Н35СООН + НS~КоА + АТФ → С17Н35СО~ SKoA + АМФ + 2Н3РО4
Стеариновая кислота Кофермент А Стеарил-КоА
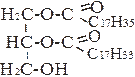 |
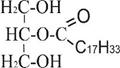
Б) + С17Н35CO ~ SKoA → + НS~KoA
2-моноацилглицерол 1,2-диацилглицерол
(2-олеоглицерол) (стеароолеоглицерол)
 | 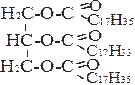 | ||
В) + С17Н35СО~ SKoA → + НS~KoA
1,2-диацилглицерол триацилглицерол
(олеодистеарин)
Следовательно, ресинтез жиров может идти непосредственно без промежуточных стадий из промежуточных продуктов гидролиза пищевых жиров – 2-моноацилглицеролов.
Однако в эпителиальных клетках тонкого кишечника содержаться фермент моноацилглицероллипаза, расщепляющая 2-моноацилглицерол на глицерол и жирную кислоту, которые подвергаются дальнейшим превращениям.
Другой путь ресинтеза жиров может протекать из продуктов полного гидролиза экзогенных пищевых жиров. Основные реакции его сходны с механизмом синтеза нейтральных жиров (см.п.2.1.), отличие заключается в том, что активируются на 2-м этапе экзогенные высшие жирные кислоты.
2.3. Биосинтез жирных кислот
С пищей в организм поступают разнообразные жирные кислоты, в том числе и незаменимые. Значительная часть заменимых жирных кислот синтезируется в печени, в меньшей степени — в жировой ткани и лактирующей молочной железе. Источником углерода для синтеза жирных кислот служит ацетил-КоА, образующийся при распаде глюкозы в абсорбтивном периоде. Таким образом, избыток углеводов, поступающих в организм, трансформируется в жирные кислоты, а затем в жиры.
Долгое время считали, что биосинтез высших жирных кислот осуществляется путем обращения реакций β-окисления высших жирных кислот. Однако в последние десятилетия было обнаружено, что для его осуществления необходим не только ацетил-Ко А, но и СО2 (из которых при посредстве АТФ-зависимой реакции возникает малонил-КоА), а сам процесс ускоряется синтетазой высших жирных кислот, локализованной в растворимой фракции клетки, эта точка зрения была отвергнута. В 60-е годы огромную роль в расшифровке механизма биосинтеза высших жирных кислот сыграли работы Ф. Линена и сотрудники.
Рассмотрим некоторые важные особенности биосинтеза жирных кислот.
1. Полный синтез de novo насыщенных жирных кислот как у прокариот, так и у эукариот осуществляется только в растворимой части цитоплазмы.
2. Промежуточные продукты синтеза жирных кислот ковалентно связаны с сульфгидрильными группами ацилпереносящего белка (АПБ).
3. Многие ферменты синтеза жирных кислот у высших организмов организованы в мультиферментный комплекс, называемый синтетазой высших жирных кислот (ВЖК).
4. Растущая цепь жирной кислоты удлиняется путем последовательного присоединения двух углеродных компонентов, происходящих из ацетил-КоА. Активированным донором двух углеродных компонентов на стадии элонгации служит малонил-АПБ. Реакция элонгации запускается высвобождением углекислого газа.
5. Роль восстановителя при синтезе жирных кислот выполняет НАДФН+Н+.
6. Элонгация под действием комплекса синтетазы жирных кислот останавливается на этапе образования пальмитата (С16).
Дальнейшая элонгация и введение двойных связей осуществляется другими ферментными системами.
Синтез жирных кислот состоит из нескольких этапов:
А. Образование ацетил-КоА и его транспорт в цитозоль.
Б. Образование малонил-КоА из ацетил-КоА.
В. Реакции, катализируемые синтетазой ВЖК (синтез пальмитиновой кислоты).
Г. Удлинение цепи и образование двойных связей в молекулах ВЖК.
А. Образование ацетил-КоА и его транспорт в цитозоль
Строительным блоком для синтеза жирных кислот служит ацетил-КоА, который в основном образуется в митохондриях за счет окисления жирных кислот, а также при дихотомическом распаде углеводов.
Так как синтез жирных кислот происходит в цитозоле клеток, то ацетил-КоА должен быть транспортирован через внутреннюю мембрану митохондрий в цитозоль. Однако внутренняя мембрана митохондрий непроницаема для ацетил-КоА, поэтому существует несколько специальных механизмов для перевода CH3CO-SKoA в транспортабельную форму.
Митохондриальный ацетил-КоА взаимодействует с оксалоацетатом, в результате образуется цитрат, который свободно проникает в цитоплазму клетки, где расщепляется до ацетил-КоА и ЩУК.

В данном случае цитрат выступает в роли переносчика ацетильного радикала. Перенос цитрата в цитоплазму происходит только при увеличении количества цитрата в митохондриях, когда изоцитратдегидрогеназа и α-кетоглутаратдегидрогеназа ингибированы высокими концентрациями НАДH и АТФ. Эта ситуация создаётся тогда, когда клетки печени получают достаточное количество источников энергии.
Есть еще один путь переноса из митохондрий ацетил-КоА в цитоплазму клетки. Это – путь с участием карнитина (витамин Вт). Выше указывалось, что карнитин играет роль переносчика ацильных групп из цитоплазмы в митохондрии при β-окислении жирных кислот. По-видимому, он может играть роль и в обратном процессе, то есть в переносе ацильных групп из митохондрий в цитоплазму; однако этот путь в синтезе жирных кислот не является главным.
Б. Образование малонил-КоА из ацетил-КоА
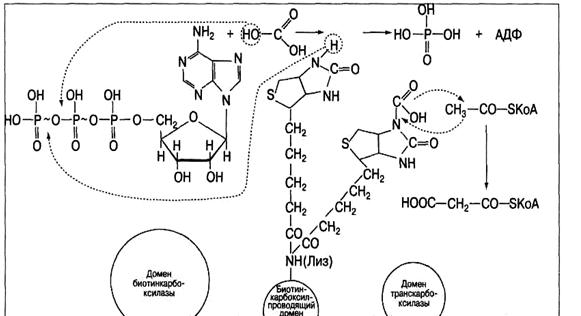
Начальный этап биосинтеза высшей жирной кислоты, приводящий к синтезу малонил-КоА ускоряется полифункциональным ферментом (мол. масса 225 кДа) ацетил-КоА-карбоксилазой, содержащей в качестве простетической группы биотин (витамин Н). Она включает в себя домен биотин-карбоксилазы, биотинкарбоксилпроводящий домен и домен транскарбоксилазы.
Эта необратимая реакция представляет собой решающий этап в синтезе жирных кислот, катализируется. Реакция идет в две стадии:
1)  биотин-фермент + CO2+H2O + АТФ карбоксибиотин-фермент + АДФ + H3PO4;
биотин-фермент + CO2+H2O + АТФ карбоксибиотин-фермент + АДФ + H3PO4;
2) карбоксибиотин-фермент + ацетил-КоА → малонил-КоА + биотин-фермент.
Суммарная реакция:
 CH3CO ~ SKoA +CO2+АТФ + H2O HCOOC-CH2CO ~SKoA+H3PO4 + АДФ
CH3CO ~ SKoA +CO2+АТФ + H2O HCOOC-CH2CO ~SKoA+H3PO4 + АДФ
Первый домен обеспечивает ускорение реакции карбоксилирования биотина (рис.12), который через радикал лизина присоединен ко второму, биотин-карбоксилпроводящему домену. Обладая высокой степенью подвижности, карбоксилированный биотин переносит СО2 в активный центр третьего домена - транскарбоксилазы, который снимает с него СО2 и непосредственно передает его на ацетил-КоА, образуя малонил-КоА.
В мономерном состоянии ацетил-КоА-карбоксилаза неактивна и приобретает способность карбоксилировать CH3CO~SKoA только после соединения мономеров в нитевидный олигомер с молекулярной массой в несколько сотен миллионов и длиной около 500 нм.
Фермент ацетил-КоА-карбоксилаза – регуляторный фермент; катализируемая этим ферментом реакция является лимитирующим этапом, определяющим скорость всего процесса биосинтеза жирных кислот в животных тканях. Главным положительным модулятором этого фермента служит цитрат, инициирующий переход фермента в высокоактивное соединение. Как только содержание цитрата в митохондриях увеличивается, что наблюдается при высокой скорости образования митохондриального ацетил-КоА и АТФ, цитрат выходит из митохондрий и выступает одновременно в роли предшественника цитозольного ацетил-КоА и аллостерического активатора ацетил-КоА-карбоксилазы.
Кроме того, активность ацетил-КоА-карбоксилазы регулируется ее фосфорилированием (снижение) и дефосфорилированием (повышение).
Присоединение новой карбоксильной группы к ацетил-КоА и необратимость реакции обеспечивается за счет энергии гидролиза АТФ.
 |
Рис.12. Механизм биосинтез малонил-КоА
В. Реакции, катализируемые синтетазой ВЖК
Биосинтез жирных кислот – циклическое повторение отдельных ферментативных реакций на мультиферментном комплексе, образованном шестью ферментами и ацилпереносящим белком (АПБ), который тоже является ферментом, находящимся в центре. Поэтому часто принимают мультиферментный комплекс (синтетазу высших жирных кислот), состоящий из семи ферментов.
Синтетаза высших жирных кислот (синтаза высших жирных кислот) - полифункциональный комплекс у высокоорганизованных форм имеет молекулярную массу 400 000-560 000 Да. Синтаза высших жирных кислот позвоночных состоит из двух идентичных полипептидных цепей, то есть представляет собой димер. Каждая из цепей (мономеров) длиной около 2300 аминокислотных остатков образует в третичной структуре три домена и в их составе 7 субдоменов, одним из которых является ацилпереносящий белок (АПБ), а шести остальным присуща определенная ферментативная функция (рис.13).
В каждый из двух мономеров в составе соответствующих доменов входят следующие ферменты (субдомены):
1) β-кетоацилсинтаза (конденсирующий фермент);
2) трансацилаза (сочетает две активности: ацилтрансферазы и малонил- трансферазы);
3) β-кетоацилредуктаза;
4) β-оксиацилдегидратаза;
5) еноилредуктаза;
6) тиоэстераза (принимает участие только на заключительной стадии синтеза пальмитиновой кислоты).
Ацилпереносящий белок (АПБ) служит «якорем», к SH-группе которого в ходе удлинения цепи жирной кислоты прикрепляются ацильные промежуточные продукты.
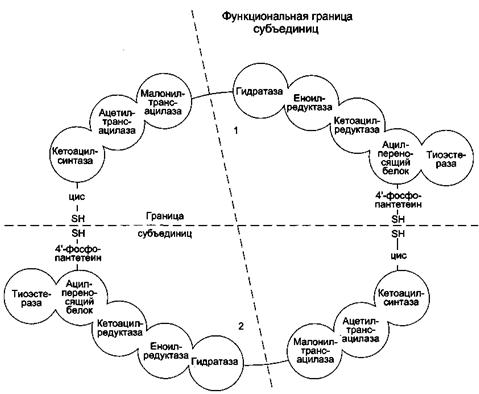
Рис.13. Синтетаза ВЖК
Примечание: Комплекс — димер из двух идентичных полипептидных цепей, каждый из которых имеет 7 активных центров и ацилпереносящий белок (АПБ). SH-группы протомеров принадлежат различным радикалам. Одна SH-группа принадлежит цистеину, другая — остатку фосфопантетеиновой кислоты. SH-группа цистеина одного мономера расположена рядом с SH-группой 4-фосфопантетеината другого протомера. Таким образом, протомеры фермента расположены «голова к хвосту». Хотя каждый мономер содержит все каталитические центры, функционально активен комплекс из 2 протомеров. Поэтому реально синтезируются одновременно 2 жирных кислоты. Для упрощения в схемах обычно изображают последовательность реакций при синтезе одной молекулы кислоты.
Специфическая SH-группа тиоламина АПБ входит в состав 4/-фосфопантотеновой простетической группы, включающей витамин В3, то есть АПБ снабжен своеобразной вращающей «ручкой», торчащей наружу из центральной части комплекса. Она перемещает промежуточные соединения от одного фермента к другому (рис.14).
4/-Фосфопантотеин
 |



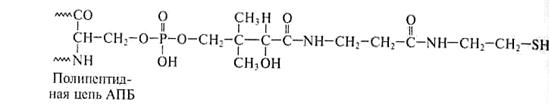
Пантотеновая кислота
Рис.14. Строение «ручки» (простетической группы) АПБ
Каждый из двух мономеров синтетазы хотя и включает все семь субдоменов, участвующих в биосинтезе жирных кислот, однако не является функциональной единицей синтазы. В состав функциональной единицы синтазы входят субдомены обоих мономеров: два одного и пять другого, что показано пунктиром на рис.13.
Синтез на мультиферментном комплексе начинается с переноса ацетильной группы CH3CO~SKoA и малонильной группы HOOC-CH2CO ~ SKoA на сульфгидрильную группу АПБ:
1)Перенос ацильного остатка из CH3CO ~ SKoA на HS-группу β-кетоацил-синтазы (Еконд) на первом (одном из двух) мономере катализируется ферментом ацетил-КоА-трансферазой (трансацилазы):
CH3CO ~ SKoA + HSАПБ → CH3CO ~ SАПБ + HS~KoA
2)На следующей стадии «ручка» АПБ второго мономера принимает на себя малонил из малонил-КоА под действием фермента малонил-КоА-трансферазы (трансацилазы):
HOOC-CH2CO ~ SKoA + HS~АПБ → HOOC-CН2-CO ~ SАПБ + HS~KoA
3)Третья стадия является стадией конденсации. Ацетильный и малонильный остатки, связанные каждый с АПБ, взаимодействуют между собой (конденсируются) с выделением углекислого газа и образованием ацетоацетил-SАПБ (β-кетобутирил-SАПБ). Реакция катализируется
β-кетоацил-АПБ-синтетазой. Реакция конденсации является высоко экзергоничной реакцией:
CH3CO~SAПБ + HOOC-CH2CO~SAПБ → CO2 + CH3COCH2CO~SAПБ + HS~AПБ β-кетобутирил-SАПБ
Ацетил-Еконд и малонил-АПБ на первом мономере реагируют друг с другом таким образом, что ацетильная группа ацетил-Еконд доставляет 3-й и 4-й атомы углерода для ацетоацетильной группы β-кетобутирил-АПБ. В результате этой реакции из свободной карбоксильной группы малонил-АПБ высвобождается СО2.
Образующаяся молекула СО2 содержит тот же атом углерода, который включился в малонил-АПБ в процессе ацетил-КоА-карбоксилазной реакции. Оксид углерода (IV) в синтезе высших жирных кислот играет роль катализатора, поскольку он удаляется сразу же после того, как встраивается каждая следующая двууглеродная единица. Декарбоксилирование малонила позволяет реакции пройти до конца и является движущей силой биосинтеза жирных кислот.
4)На четвертой стадии β-кетобутирил-АПБ восстанавливается. Фермент β-кетоацил-АПБ-редуктаза, коферментом которого является восстановленный НАДФН+Н+, превращает β-кетобутирил-АПБ в β-гидроксибутирил-АПБ (β-оксибутирил-АПБ):
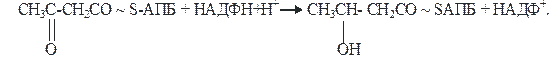
β-кетобутирил-АПБ β-гидроксибутирил-АПБ
5)На пятой стадии образующийся β-оксибутирил-АПБ дегидратируется на втором мономере. Дегидратация приводит к образованию ненасыщенного кротонил-АПБ (еноил-АПБ) при участии фермента β-гидроксиацил-АПБ-дегидратаза:
 CH3CH-CH2-CO~S-АПБ → СH3CH=CHCO~S-АПБ + H2O.
CH3CH-CH2-CO~S-АПБ → СH3CH=CHCO~S-АПБ + H2O.
OH
β-гидроксибутирил-АПБ кротонил-АПБ
6)На шестой стадии происходит восстановление ненасыщенного кротонил-АПБ до бутирил-АПБ, которое катализируется ферментом еноил-АПБ-редуктазой, коферментом которого является НАДФН+Н+:
CH3CH=CH-CO~S-АПБ + НАДФН+Н+ → НАДФ+ + CH3CH2CH2CO ~S-АПБ.
кротонил-АПБ бутирил-АПБ
Первый из серии циклов заканчивается образованием бутирил-АПБ. Перед вторым циклом радикал бутирила переносится из позиции 2 в позицию 1 (где находился ацетил в начале первого цикла реакций). Затем остаток бутирила подвергается тем же превращениям (начиная с реакции конденсации) и удлиняется на 2 углеродных атома, происходящих из малонил-КоА. В каждом цикле малонил-S-АПБ связывается с концевым углеродным атомом растущей цепи жирной кислоты с одновременным высвобождением CO2 и HS~АПБ при действии фермента β-кетоацил-АПБ-синтетазы.
Аналогичные циклы реакций повторяются до тех пор, пока не образуется радикал пальмитиновой кислоты, который под действием тиоэстеразного центра гидролитически отделяется от ферментного комплекса, то есть происходит деацилирование – высвобождение свободной пальмитиновой кислоты при действии гидролитического фермента ацилгидролазы (рис.15).
В каждом цикле биосинтеза пальмитиновой кислоты проходят 2 реакции восстановления, донором водорода в которых служит кофермент НАДФH, восстановление которого происходит в реакциях:
• дегидрирования в окислительных стадиях пентозофосфатного пути катаболизма глюкозы;
• дегидрирования малата;
•дегидрирования изоцитрата цитозольной НАДФ-зависимой дегидрогеназой.
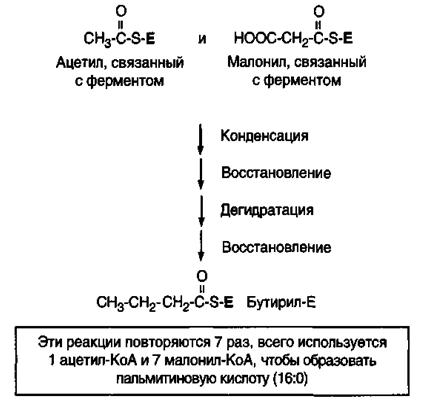
Рис.15. Общая схема реакций синтеза пальмитиновой кислоты
Суммарное уравнение реакции образования пальмитиновой кислоты при участии синтазы имеет следующий вид:
Ацетил-КоА + 7 Малонил-КоА + 14 НАДФН + 14Н+ →
→ СН3(СН2)14СООН + 7СО2 + 8НS~КоА + 14 НАДФ+ + 6Н2О
Для образования жирной кислоты с n числом атомов углерода, необходимо пройти (n/2 - 1) циклов.
Объединение всех ферментов синтеза высших жирных кислот в единый полиферментный ансамбль обеспечивает высокую эффективность работы синтазы: одновременно в пределах одного димера образуются две молекулы высшей жирной кислоты.
Основные стадии биосинтеза высших жирных кислот в организме представлены на рис. 16.
Долгое время считалось, что печень является единственным органом, где происходит синтез жирных кислот (ЖК). В настоящее время установлено, что синтез ЖК имеет место также в стенке кишечника, в легочной ткани, в жировой ткани, в ткани мозга, в почках, в костном мозге, в лактирующей молочной железе и даже в сосудистой стенке. Он протекает в цитозоле клетки. Характерно, что в цитозоле печеночных клеток синтезируется главным образом пальмитиновая кислота.
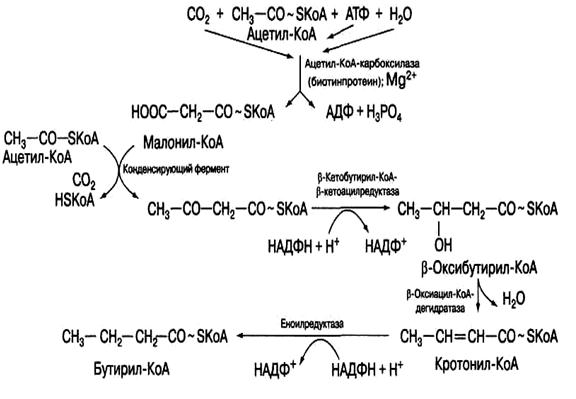
Рис.16. Механизм биосинтеза высших жирных кислот
Г. Удлинение цепи и образование двойных связей в молекулах ВЖК
Удлинение жирных кислот на основе пальмитиновой кислоты проиходит с участием малонил-КоА. Последовательность реакций сходна с той, что происходит при синтезе пальмитиновой кислоты; однако в данном случае жирные кислоты связаны не с синтетазой жирных кислот, а с коферментом А. Ферменты, участвующие в элонгации, могут использовать в качестве субстратов не только пальмитиновую, но и другие жирные кислоты; поэтому в организме могут синтезироваться не только стеариновая кислота, но и жирные кислоты с большим числом атомов углерода.
Основной продукт элонгации в печени - стеариновая кислота
| <== предыдущая страница | | | следующая страница ==> |
| Фосфатидилинозитол | | | А. Нарушение процессов всасывания жиров |
Дата добавления: 2014-10-02; просмотров: 1775; Нарушение авторских прав

Мы поможем в написании ваших работ!