Главная страница Случайная лекция

Мы поможем в написании ваших работ!
Порталы:
БиологияВойнаГеографияИнформатикаИскусствоИсторияКультураЛингвистикаМатематикаМедицинаОхрана трудаПолитикаПравоПсихологияРелигияТехникаФизикаФилософияЭкономика

Мы поможем в написании ваших работ!
Лекция 4
Классификация органических соединений.
Все органические соединения в зависимости от строения углеродной цепи – скелета – делят на следующие группы.
1. Алициклические соединения – соединения с открытой незамкнутой цепью углеродных атомов (прямой или разветвленной)
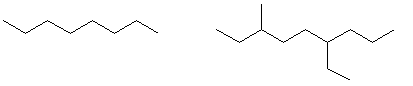
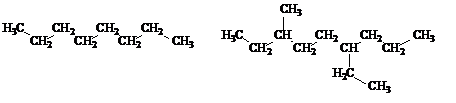
Такие соединения называются алифатическими соединениями или соединениями жирного ряда.
2. Циклические соединения – соединения, в которых углеродные атомы образуют циклы. Эти соединения делят на карбоциклические и гетероциклические.
А) Карбоциклические (изоциклические) – циклические соединения, образованные только углеродными атомами. В свою очередь карбоциклические соединения могут быть
а) алициклическими
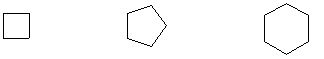
Циклобутан Циклопентан Циклогексан
и б) ароматическими.
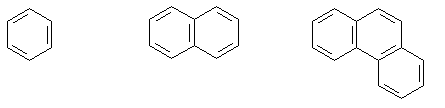
Бензол Нафталин Фенантрен
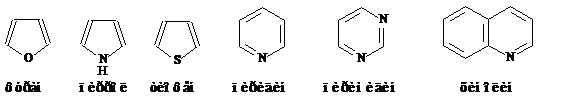
Б) Гетероциклические соединения – циклические соединения, в состав цепи которых входят и другие атомы – гетероатомы.
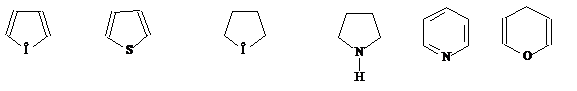
Фуран тиофен тетрагидрофуран пирролидин пиридин γ-пиран
Наиболее простыми представителями соединений ациклического, алициклического и ароматического рядов являются углеводороды, молекулы которых состоят только из двух элементов: С и Н. С этих соединений обычно начинают изучение органической химии.
При замене атомов водорода в углеводородах на другие атомы или группы атомов – функциональные группы – образуются многочисленные классы органических соединений.
Углеводороды R−H Сложные эфиры R−COOR
Галогенопроизводные R−Hal Нитросоединения R−NO2
Спирты R−OH Аминосоединения R−NH2
Альдегиды R−CHO Сульфокислоты R−SO3H
Кетоны R−CO−R Металлоорганические соединения
Кислоты R−COOH R−Me, R−Me−Hal
Простые эфиры R−O−R
Из этого ряда видно, что функциональная группа определяет класс соединения. Органические вещества, содержащие две или более различные функциональные группы, называются соединениями со смешанными функциями.
Каждый из этих классов делится в зависимости от характера связи между углеродными атомами на две группы: предельные и непредельные.
Номенклатура органических соединений
Тривиальная номенклатура
Это система исторически сложившихся названий, но применяемых до настоящего времени. Как правило, эти названия были даны еще в ранний период развития органической химии и никак не отражают строение органического вещества.
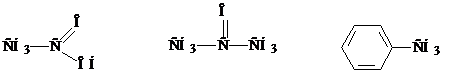
уксусная кислота ацетон толуол
Рациональная номенклатура
По правилам рациональной номенклатуры за основу названия органического соединения принимают название наиболее простого (чаще всего первого) члена данного гомологического ряда Все остальные соединения рассматриваются как его производные, образованные замещение в нем атомов водорода алкильными группами, атомами или функциональными группами. Названия алкильных и функциональных групп, наиболее часто встречающихся в структурных формулах органических молекул, приведены в таблице 1 и 2.
Таблица 1.
Названия некоторых алкильных групп
| Структурная формула группы | Название (краткое обозначение) | Структурная формула группы | Название (краткое обозначение) |
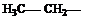
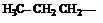
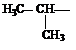
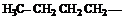
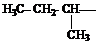
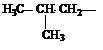
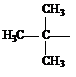
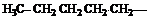
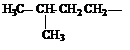
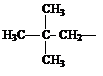
| Метил (Ме) Этил (Еt) Пропил (Pr) Изопропил (i-Pr) Бутил (Bu) втор-Бутил (s-Bu) Изобутил (i-Bu) трет-Бутил Пентил (амил) Изопентил (изоамил) Неопетил | 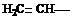
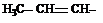
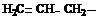
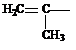
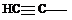
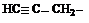
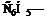
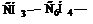
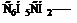
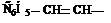
| Винил, этинил Пропенил Аллил Изопропенил Этинил Пропаргил Фенил (Ph) Толил (о-, м-, п-) Бензил (Bn) Стирил |
Таблица 2.
Названия органических соединений некоторых классов
по рациональной номенклатуре
| Класс | Соединение – основа названия | Примеры |
| Насыщенные углеводороды Ненасыщенные углеводороды Ацетиленовые углеводороды Спирты Альдегиды Кислоты | СН4
метан
СН2=СН2
этилен
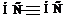 ацетилен
СН3ОН
карбинол
ацетилен
СН3ОН
карбинол
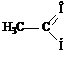 уксусный альдегид
уксусный альдегид
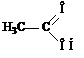 уксусная кислота
уксусная кислота
| 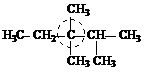 диметилэтилизопропилметан
диметилэтилизопропилметан
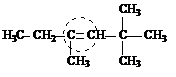 α-метил-α-этил-β-трет-бутилэтилен
α-метил-α-этил-β-трет-бутилэтилен
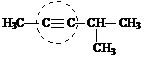 метилизопропилацетилен
метилизопропилацетилен
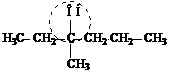 метилэтилпропилкарбинол
метилэтилпропилкарбинол
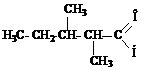 метил-втор-бутилуксусный альдегид
метил-втор-бутилуксусный альдегид
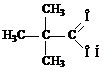 триметилуксусная кислота
триметилуксусная кислота
|
Систематическая номенклатура ИЮПАК
Для того чтобы назвать органическое соединение по систематической номенклатуре ИЮПАК, нужно:
1. выбрать родоначальную структуру;
2. выявить все имеющиеся в соединении функциональные группы;
3. выбрать, какая группа является старшей (таблица 3); название этой группы отражается в названии соединения в виде суффикса и его ставят в конце названия соединения; все остальные группы дают в названии в виде префиксов (приставок);
4. обозначить ненасыщенность соответствующим суффиксом (-ен или -ин), а также префиксом (дегидро-, тетрагидро- и др.);
5. пронумеровать главную цепь, придавая старшей группе наименьший из номеров;
6. перечислить префиксы (приставки) в алфавитном порядке (при этом умножающие префиксы ди-, три- и т.д. не учитываются);
7. составить полное название соединения
Таблица 3
Наиболее важные функциональные группы, которые могут быть представлены в названиях органических соединений префиксами и суффиксами
(приведены в порядке убывания старшинства)
| Название класса | Формула группы | Название группы в виде префикса | Название группы в виде суффикса |
| Катионы Карбоновые кислоты Сульфокислоты Производные кислот: ангидриды сложные эфиры галогенангидриды амиды нитрилы Альдегиды Кетоны Спирты Тиолы Амины Имины Простые эфиры Сульфиды Галогенпроизводные Нитрозопроизводные Нитропроизводные | 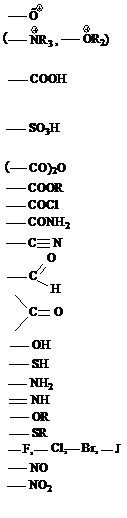
| онио- карбокси- сульфо- − алкоксикарбонил- хлорформил- карбамоил- циан- формил- оксо- гидрокси- меркапто- амино- имино- алкокси- алкилтио- галоген- нитрозо- нитро- | -оний -овая кислота (карбоновая кислота) -сульфонокая -ангидрид -оат -оилхлорид -амид -нитрил -аль -он -ол -тиол -амин -имин -оксид (эфир) -сульфид -галогенид − − |
Родоначальная структура – главная цепь а ациклической молекуле или гетероциклическая система (или ее часть), лежащая в основе соединения.
В ациклических соединениях главной цепью называют цепь углеродных атомов, составляющую основу названия и нумерации. В состав этой цепи обязательно должна входить старшая характеристическая группа. Главная цепь должна содержать наибольшее число заместителей, максимальное число кратных (двойных и тройных) связей и должна быть наиболее длинной.
Главную цепь нумеруют в соответствии с наименьшей суммой цифровых индексов, указывающих положения заместителей и кратных связей.
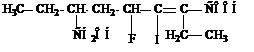
7-гидрокси-3-иод-4-фтор-2,6-диэтил-2-гептеновая кислота
(в этой нумерации в главную цепь включены старшая группа СООН и один из заместителей - ОН)
В случае кратных связей при одинаковых цифровых индексах двойных и тройных связей предпочтение отдается двойной связи.
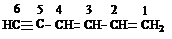
1,3-гексадиен-5-ин
В алициклических соединениях главной цепью считают замкнутую цепь углеродных атомов в названии этой цепи применяют префикс цикло-.
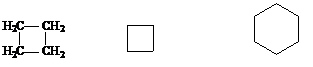
циклобутан циклогексан
В основе названий ароматических соединений лежат тривиальные названия.
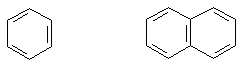
бензол нафталин
В гетероциклических соединения за основу берут название соответствующего гетероцикла.
При выборе старшей группы важно старшинство заместителей относительно друг друга.

старшая старшая
группа группа
3-гидроксипропановая кислота 3-нитро-1-пропанол
После того как определена родоначальная структура, выбрана старшая группа (обозначается суффиксом), проведена нумерация, выявлены другие группы, составляют полное название. При этом цифровые индексы (локанты), указывающие положение заместителей и кратных связей, дают перед префиксом и перед суффиксом.

Углеводороды алифатического ряда.
Все алифатические углеводороды можно разделить по характеру связей между атомами углерода на две большие группы: предельные (насыщенные) и непредельные (ненасыщенные) углеводороды.
Насыщенные углеводороды (алканы, парафины).
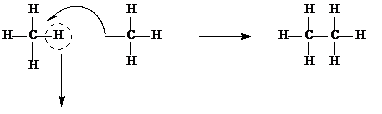
Строение алканов. Гомологический ряд. Радикалы. Простейшим представителем предельных углеводородов является метан СН4. Атом углерода имеет sp3-гибридизацию, угол между связями 109° 28’. Заменяя атом водорода на метильную группу можно получить структурную формулу следующего за метаном углеводорода – этана С2Н6.
Добавляя следующую метильную группу, получим следующий за метаном углеводород – пропан С3Н8.
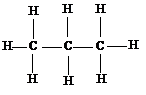
Таким образом, можно вывести формулы остальных представителей предельных углеводородов. Все предельные углеводороды объединяют в ряд в соответствии с ростом числа атомов углерода цепи, причем по составу углеводороды отличаются по составу на группу СН2, называемую гомологической разностью. Общая формула гомологического ряда предельных углеводородов имеет вид СnН2n +2.
Если углеводород лишается одного или нескольких атомов водорода, то образуется структура называется углеводородным радикалом. (метил, метилен, метин). Если свободная валентность находится у атома углерода, соединенного с тремя атомами водорода это метильный радикал, с двумя атомами водорода и одним углерода то это первичный радикал. Если у атома углерода одна связь с водородом, а две с углеродом, то это вторичный радикал. Если свободная валентность находится у атома углерода, соединенного с тремя атомами углерода, то это третичный радикал. Название радикала получают заменой окончания ан в названии углеводорода на ил (например этан – этил , бутан – бутил и.т.д.).
Номенклатура и изомерия.
Для названия углеводородов используют в основном номенклатуру IUPAC.
Общее название углеводородов – алканы.
1. В молекуле углеводорода выбирают наиболее длинную цепь.
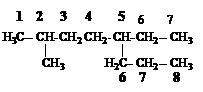
2. Цепь нумеруют таким образом, чтобы сумма номеров заместителей была минимальна. Если в цепи несколько одинаковых заместителей, то перечисляются номера заместителей, а затем греческими числительными указывают число одинаковых заместителей.
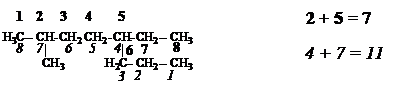
3. Если в качестве основной цепи можно выбрать несколько разных с одинаковой длиной, то из них выбирают такую цепь, которая имеет максимальное число заместителей.
3,4,5-триметил-4-бутилоктан, а не 3-метил-4-(1’,2’-диметилбутил)октан
Изомерия. Для алканов характерна структурная изомерия. Изомеры – это соединения, имеющие одинаковый состав, но разное химическое строение.
При названии разветвленных углеводородов часто используют приставку изо-. Например: н-бутан и изо-бутан.

В первом случае все четыре связи образуют прямую (нормальную) цепь, а втором случае – разветвленную (изостроение). Необходимо отметить, что в углеводородах даже с прямой цепью атомы углерода располагаются не по прямой, а имеют зигзагообразную форму с межвалентными углами 109о 28’. Соединения с прямой цепью называют нормальными (обозначают буквой н-), а с разветвленной (приставка изо-).
При выводе структурных формул отдельных изомеров можно поступать следующим образом.
1. В соотвествии с числом углеродных атомов в молекуле записывают прямую углеродную цепь (углеродный скелет). Например С5Н12 состоит из пяти атомов углерода:
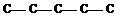
1 2 3 4 5
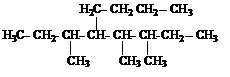
2. «Отщепляя» крайние углеродные атомы располагают их у оставшихся в цепи, добиваясь максимально возможного числа перестановок. При этом первоначальная углеродная цепь укорачивается, структура становится более разветвленной.
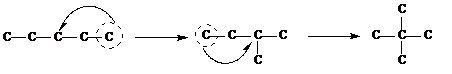
Необходимо помнить, что «изогнув» произвольно молекулу, нельзя получить новый изомер. Например:
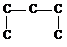
3. Соблюдая условия четырехвалентности углеродных атомов, заполняют оставшиеся валентности атомами водорода.
Чем больше в молекуле атомов углерода, тем больше изомеров. В зависимости от положения в углеродной цепи атом углерода может быть первичным (если углерод затрачивает одну валентность на связь с другим углеродным атомом), вторичным (если соединен с двумя другими углеродными атомами), третичным (связан с тремя другими атомами углерода), четвертичным (связан с четырьмя другими углеродными атомами).
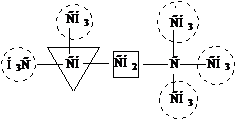
Здесь первичные атомы углерода заключены в кружок, вторичный атом – в квадрат, третичный – в треугольник, четвертичный – в пунктирный кружок.
Лекция 5
Способы получения алканов.
Природные источники. Для получения предельных углеводородов используют в основном природные источники (газ, нефть, уголь, древесину, торф, и др.) и синтетические методы. Например, метан, этан, пропан и другие находятся в природном газе или растворены в нефти. Метан образуется при действии анаэробных (развивающихся без доступа воздуха) микробов на различные органические остатки (например, целлюлозу):
(С6Н10О5)n + nН2О → 3nCO2 + 3nСН4
Алканы можно получать гидрированием углей, сухой перегонкой углей, древесины, торфа (нагревание без доступа воздуха), перегонкой нефти.
Синтетические методы.
1. Гидрирование непредельных углеводородов (этилен, ацетилен) в присутствии катализатора.
С2Н4 + Н2 → С2Н6
2. Синтез Вюрца (1870). Метод позволяет получать симметричные алканы с большим содержанием атомов углерода.
СН3 – I + Na + I – CH3 → CH3 – CH3 + 2NaI
Если в реакции используется два галогеналкила (СН3I и C2H5I), то образуется не один продукт, а смесь (СН3 – СН3, С2Н5 – С2Н5, СН3 – С2Н5).
3. Синтезы на основе Mg органических соединений.
R1Br + Mg → R1MgBr
R1MgBr + R2Br → R1 – R2 + MgBr2
4. Щелочное плавление карбоновых кислот.
СН3 – СООNa + NaOH → СН4 + Na2CO3
Физические свойства. Первые четыре члена гомологического ряда – газы, С5 – С15 – жидкие, С16 и выше – твердые. Алканы легче воды и в ней практически не растворяются, но растворяются в большинстве органических растворителей. Жидкие алканы – хорошие растворители для многих органических веществ. Температуры кипения и плавления приводятся в справочниках и учебниках (Артеменко, 2000, стр. 56.)
Химические свойства. В молекулах углеводородов все атомы связаны прочными σ-связями, валентности углеродных атомов полностью насыщены водородом. Потому предельные углеводороды проявляют высокую химическую инертность: не вступают в реакции присоединения, не взаимодействуют с ионными реагентами (кислотами и щелочами), окислителями, активными металлами.
Основные превращения предельных углеводородов идут только при воздействии высокой энергии (нагревание, УФ-облучение). При этом может произойти разрыв связи С – Н с заменой водорода на другой атом или группу атомов, или разрыв связи С – С. Хотя энергия связи С – Н больше энергии связи С – С, разрыв идет преимущественно по связи С – Н, поскольку эти связи более доступны для атаки химическими реагентами.
Связи С – С и С – Н, обладают незначительной полярностью, поэтому при разрыве образуют в основном не ионы, а радикалы, т.е. разрыв протекает по гомолитическому механизму. Таким образом, для предельных углеводородов характерны два основных типа реакций:
1. реакции замещения водорода с разрывом С – Н связи;
2. реакции расщепления молекулы с разрывом как С – Н, так и С – С связей.
Реакции замещения. В этих реакциях легче всего происходит замещение атома водорода, связанного с третичным атомом, труднее – со вторичным. Это объясняется величинами энергий связей С – Н у первичного (около 420 кДж/моль), вторичного (390 кДж/моль) и третичного (370 кДж/моль).
Основные реакции замещения предельных углеводородов.
1. Галогенирование. Реакция идет при облучении или нагревании.
СН4 + Cl2 → CH3Cl + HCl.
Реакция протекает по цепному радикальному механизму, включающему стадии инициирования, роста и обрыва цепи.
2. Нитрование по Коновалову (12 – 14% р-р НNO3, 140 °С).
СН3 – СН2 – СН2 – СН3 + HNO3 → CH3 – CH NO2 – CH2 – CH3
2-нитробутан
Механизм цепной радикальный.
3. Сульфохлорирование и сульфоокисление (Рид и Хорн 1936).
R – H + SO2 + Cl2 → R – SO2Cl + HCl
Алкилсульфохлорид
Механизм цепной радикальный, катализатор hυ, перекиси.
R – H + Cl˙ → R˙ + HCl
Cl2 (рост цепи)
R˙ + SO2 → R – SO2˙ ―→ R – SO2Cl + Cl˙
Этим методом получают сульфокислоты с 12 – 18 атомами углерода и выше, применяемые для получения ПАВ (R – SO3 Na).
Реакции расщепления
1. Отщепление водорода (дегидрирование). Реакция протекает в присутствии катализатора (Cr2O3) при нагревании с образованием непредельных углеводородов.
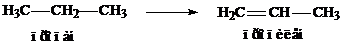
2. Реакции расщепления молекулы алкана за счет разрыва С – С связи.
Эти реакции протекают при крекинге нефти и окислении алканов.
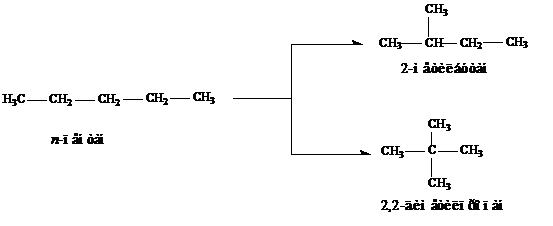
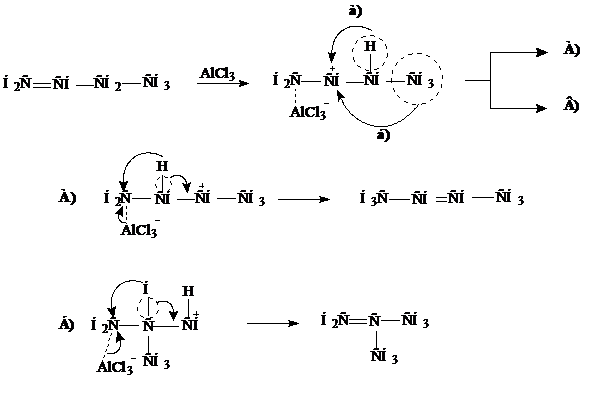
Реакции изомеризации. При нагревании алканов с числом атомов углерода не менее четырех в присутствии катализатора (АlCl3) происходит превращение неразветвленной цепи в разветвленную
Непредельные (ненасыщенные) углеводороды.
Алкены.
Алкенами или олефинами называют углеводороды, в молекулах которых между углеродными атомами имеется двойная связь. Алкены образуют гомологический ряд с общей формулой СnH2n. Простейшим представителем этого ряда является этилен.
Электронное строение и характер гибридизации двойной связи были рассмотрены ранее.
Номенклатура Согласно номенклатуре IUPAC названия алкенов производят из названий соответствующих алканов заменой суффкса ан на ен.
Например: алкан – алкен. Главная цепь должна обязательно содержать двойную связь. Цепь нумеруют таким образом, чтобы номер первого углеродного атома при двойной связи был минимальным. Цифру, обозначающую положение двойной связи, ставят перед названием основной цепи. В отечественной литературе (например, учебник Артеменко) согласно систематической номенклатуре ставят после названия цепи. В остальном порядок составления названия такой же как для алканов, соответствует правилам IUPAC
2,5-диметилгексен-2 или 2,5-димети-2-гексен
Алканы простого строения часто называют, заменяя суффикс ан на илен: этан – этилен, пропан – пропилен.
Изомерия. Для алкенов кроме изомерии углеродного скелета, характерна изомерия положения двойной связи, а также изомерия, связанная с невозможностью вращения вокруг двойной связи без ее разрыва. Это приводит к появлению геометрических изомеров или цис- транс-изомеров. Геометрическая изомерия является одним из видов пространственной изомерии.
Изомеры, у которых заместители расположены по одну сторону от двойной связи, называются цис-изомерами, а по разные стороны – транс-изомерами. Цис- и транс-изомеры отличаются не только пространственным строением, но и многими физическими и химическими свойствами.
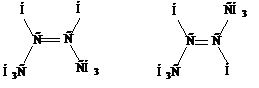
цис-бутен-2 транс-бутен-2
Способы получения алкенов
1. Крекинг нефти – термическая обработка нефти и нефтепродуктов.
2. Дегидрогенизация алканов в присутствии катализаторов при 560 – 620 °С.
3. Дегидратация спиртов в присутствии кислотных катализаторов Н2SO4, H3PO4, Al2O3 и др.
140°C, кат. 160-170°C
СН3СН2ОН + Н2SO4 → СН3СН2О-SO2-OH → CH2=CH2
-H2O -H2SO4
При дегидратации спиртов атом водорода отщепляется от наименее гидрогенизированного атома углерода, т.е. вторичного или третичного (правило Зайцева)
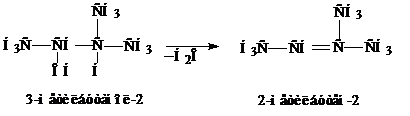
4. Дегидрогалогенирование алкилгалогенидов под действием спиртового или твердого КОН.
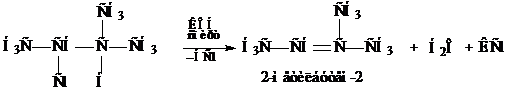
Отщепление НСl проходит по правилу Зайцева. Механизм таких реакций, называемых реакциями отщепления, рассмотрим позднее. Здесь отметим, что чем устойчивей алкен, тем легче он образуется. Наиболее замещенные алкены наиболее устойчивы.
5. Дегалогенирование дигалогенпроизводных с атомами галогена у соседних атомов углерода под действием Zn пыли в спирте.
R – CHCl – CH2Cl + Zn → R – CH2 = CH2 + ZnCl2
6. Селективное гидрирование в присутствие катализатора
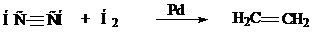
Физические свойства (Самостоятельно Артеменко)
Химические свойства.
Реакции присоединения
1. Гидрирование. Присоединение водорода происходит в присутствии катализатора (Pt или Pd) при нормальной температуре.
СН2=СН2 + Н2 → СН3 – СН3
Реакционная способность алкенов меняется в соответствии с правилом С.В.Лебедева: алкены гидрируются тем легче, чем меньше заместителей содержится у двойной связи.
2. Присоединение галогенов (галогенирование).
СН3 – СН = СН2 + Cl2 → СН3 – СНСl – СН2Cl
Реакционная способность галогенов Cl2 > Br2 > I2.
Присоединение галогенов в зависимости от условий может протекать как по ионному так и по радикальному механизму. В первом случае под влиянием двойной связи происходит поляризация молекулы галогена и поляризованная молекула образует неустойчивую промежуточную частицу – π-комплекс
δ− δ+ СН2 СН2+ CH2Cl CH2Cl
Сl – Cl ← ∙∙∙∙ || | | Сl− |
СН → СНСl + CH+ → CHCl
π-комплекс | | | |
СН3 CH3 CH3 CH3
I II
карбокатион
Карбокатион II более устойчив, в его стабилизации больше донорных заместителей (СН3 – группа), поэтому он образуется в большей степени.
Под действием света или в присутствии перекисей реакция присоединение галогенов может носить радикальный характер, поскольку молекула галогена в этих условиях распадается на два радикала (атакующая частица – радикал)
СН2 СН2˙ CH2Br CH2Br
|| | | Br˙ |
СН + Вr˙ → СНBr + CH˙ → CHBr
| | | |
СН3 CH3 CH3 CH3
I II
Радикал II более устойчив, чем I, поскольку действует правило: более замещенный алкильный радикал более устойчив. Этому есть объяснение, но более детально причины мы рассматривать не будем.
3. Гидрогалогенирование (присоединение галогеноводородов)
СН3 – СН – СН2Br
1-бромпропан
СН3 – СН = СН2 + НBr →
СН3 – СНBr – СН3
2-бромпропан
При ионном присоединении галогеноводородов к несимметричным алкенам водород присоединяется по месту двойной связи к наиболее гидрогенизованному (связанному с наибольшим числом атомов водорода) атому углерода, а галоген – к наименее гидрогенизированному. Это правило Марковникова.
Механизм присоединения объясняет наблюдаемую ориентацию. Реакция идет в две стадии.
δ− δ+ СН2 СН2+ CH3 CH3
Br – Н ← ∙∙∙∙ || | | Br− |
СН → СН2 + CH+ → CHBr
π-комплекс | | | |
СН3 CH3 CH3 CH3
I II
карбокатионы
Карбокатион П более устойчив, образуется в большей степени, и это обстоятельство определяет ориентацию присоединения (правило Марковникова). Образуется в основном 2-бромпропан.
Правило Марковникова можно сформулировать следующим образом: Присоединение протона к алкену происходит с образованием наиболее устойчивого карбокатиона.
Присоединение НBr к пропену в присутствии перекисей (перекисный эффект Караша) приводит к образованию в основном 1-бромпропана. Причина в том, что реакция проходит по радикальному механизму, атакующей алкен частицей является не Н+ а Br˙ и более устойчивым является более замещенный радикал П.
R – CO – O – O – CO – R → 2R – COO˙
R – COO˙ + HBr → R – COOH + Br˙
СН2 СН2˙ CH2Br CH2Br
|| | | HBr |
СН + Вr˙ → СНBr + CH˙ → CH2 + Br˙
| | | |
СН3 CH3 CH3 CH3
I II
Далее реакция идет по цепному радикальному механизму. Перекисный эффект характерен только в случае присоединения HBr.
4. Гидратация. В присутствии кислотных катализаторов (Н2SO4, ZnCl2 и др.) к алкенам присодиняется вода с образованием спиртов. Присоединение воды идет по правилу Марковникова.
Н2SO4
СН3 – СН = СН2 + Н – ОН → СН3 – СНОН – СН3
5. Алкилирование алканов Алкилирование алканов – реакция с помощью, которой можно вводить различные углеводородные радикалы в молекулы органических соединений.
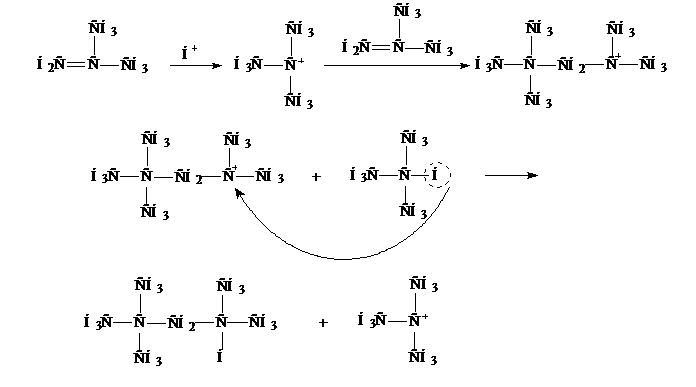
Образовавшийся третичный карбокатион вновь реагирует с алкеном и процесс повторяется циклически с образованием алкана и третичного карбокатиона
Изомеризация алкенов. При нагревании, или в присутствии катализаторов (АlCl3) алкены способны изомеризоваться. Происходит перемещение двойной связи или изменение углеродного скелета. Причина - перегруппировка промежуточно образующегося катиона с 1,2- алкильным или гидридным сдвигом.
Реакции полимеризации.
Реакция полимеризации открыта А.М. Бутлеровым. С помощью этой реакции получают различные полимеры. Эти реакции будут рассмотрены в разделе Полимеры
Реакции окисления.
1. Горение. Продукты реакции вода и СО2.
2. Образование карбоновых кислот. Окислители: кислый раствор КМnO4, хромовая смесь, НNO3(конц.)
[O]
R – CH = CH – R → 2R – COOH
3. Образование гликолей. Окислитель – разбавленный р-р КМnО4
3CH2 = CH2 + 2KMnO4 + 4H2O → 3CH2OH – CH2OH + 2MnO2 + 2KOH
Этиленгликоль
4. Образование альдегидов и кетонов. Окислитель – озон О3.
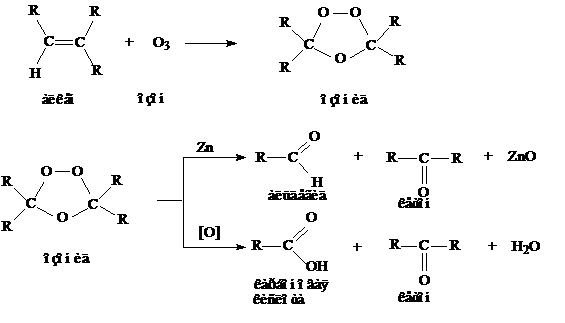
Лекция 6
Диеновые углеводороды.
Непредельные соединения, содержащие в молекуле две двойные связи, называют диеновыми углеводородами. Их называют также алкадиенами или диолефинами. Общая формула СnH2n-2.
Номенклатура. Название получается заменой суффикса ан на диен. Цепь нумеруют таким образом, чтобы цифры при двойной связи имели меньший порядковый номер. Двойные связи в цепи могут располагаться рядом, могут быть разделены σ-связью или несколькими σ-связями.
СН3 – С = С = СН2 СН2 = С – СН = СН2 СН2 = СН – СН2 – СН = СН2
| |
СН3 СН3
3-метилбутадиен-1,2 (А) 2-метилбутадиен-1,3 (В) пентадиен-1,4 (С)
(несим. диметилаллен) (изопрен)
Если связи расположены рядом, то они называются кумулированными или алленовыми: СН2=С=СН2 (пропадиен или аллен).
Если две двойные связи разделены σ-связью то их называют сопряженными
(В), если несколькими простыми связями (С) - изолированными.
Химические свойства диенов (А), (В) отличаются друг от друга от (С) для которого характерны свойства простых алкенов.
Способы получения.
1. Пиролиз нефти и нефтепродуктов.
2. Каталитическое дегидрирование н-бутана или бутилена при 600 °С на хромоалюминиевом катализаторе (оксид хрома на оксиде алюминия).
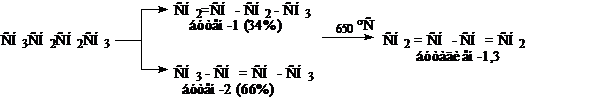
3. Дегидратация гликолей
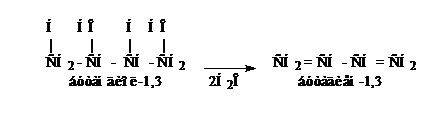
Аналогично можно получить изопрен.
4. Реакция Фаворского. Получение изопрена из ацетона и ацетилена
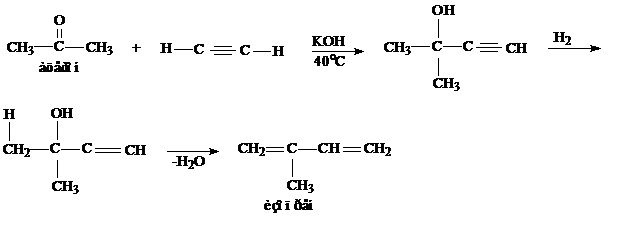
Физические свойства. Самостоятельно (Артеменко стр. 90)
Химические свойства. Диены с изолированными двойными связями ведут себя как обычные алкены. В то же время диены с сопряженными связями отличаются высокой реакционной способностью по сравнению с диенами, содержащими изолированные и кумулированные двойные связи. Эти соединения присоединяют реагент не только по одной или двум связям (1,2-присоединение), но и к противоположным концам молекулы (1,4-присоединение):
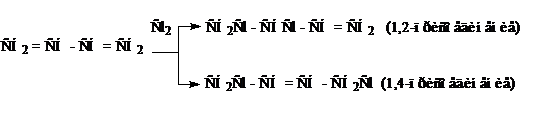
Выход продуктов 1,2- или 1,4-присоединения определяется характером реагента и условиями проведения реакции. Например, присоединение НВr в присутствии перекисных соединений идет в 1,4-положение, а в отсутствии – в положение 1,2. Водород в момент выделения (получаемый при действии кислот на металлы) присоединяется в положение 1,4, а водород в присутствии катализатора (Ni) присоединяется в положение 1,2. Продукты 1,2 присоединения подчиняются кинетическому контролю (получаются продукты скорость образования которых выше), а продукты 1,4-присоединения – продукты термодинамического контроля (образуются более устойчивые продукты). Причина – электронное строение сопряженных диенов.
Было установлено, что в бутадиене-1,3 двойные связи несколько длиннее, чем двойная связь в этилене.
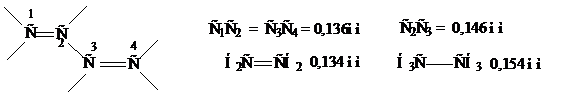
В то же время простая С – С связь короче обычной σ-связи (0,136 и 0,146 нм по сравнению с 0,134 и 0,154 нм изолированной двойной и простой С – С связи). Кроме того, молекула бутадиена плоская, т. е. все ее атомы расположены в одной плоскости.
Все это говорит о том, что в молекуле бутадиена нет «чистых» двойных и одинарных связей, а наблюдается равномерное распределение π-электронной плотности по всей молекулу с образованием единой молекулярной орбитали. Система сопряженных двойных связей ведет себя не как сумма изолированных двойных связей, а как единое целое, эффективно передающее взаимное влияние атомов. Схематично это можно представить следующим образом:
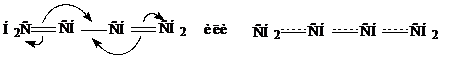
Рассмотрим механизм присоединения к НСl бутадиену-1,3. Реакция начинается с электрофильной атаки протоном π-электронного облака одной из двойных связей. В результате образуется неустойчивый π-комплекс (I), который затем превращается в сопряженный карбокатион (П). При этом π-электроны двойной связи в результате взаимодействия с положительным зарядом перемещаются в центр молекулы, это приводит к смещению заряда на крайний углеродный атом с образованием карбокатиона (Ш). Структуры (П) и (Ш) очень легко и быстро перестраиваются друг в друга. Эти Структуры называют граничными и применяют в данном случае как способ изображение мезомерного карбокатиона. Атомы С2 и С4 несущие частичный положительный заряд подвергаются затем нуклеофильной атаке со стороны Сl−, с образованием продуктов 1,2- и 1,4-присоединения:
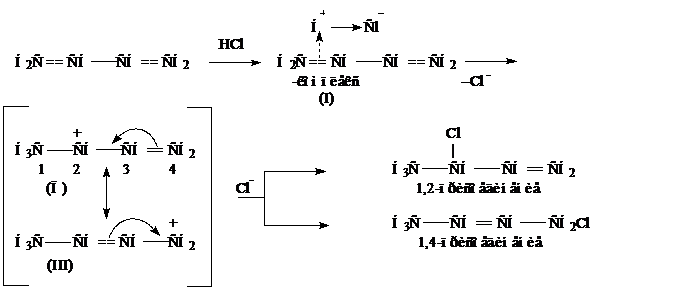
Диеновый синтез (Реакция Дильса – Альдера). Этот вид реакций состоит в 1,4-присоединении алкена или алкина к диену с сопряженными двойными связями. Простейший пример – взаимодействие бутадиена -1,3 с этиленом с образованием циклогексена.
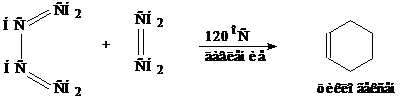
Особенно легко протекают реакции с непредельными соединениями, содержащими электроноакцепторные группы (–NO2, −C≡N, −CHO).
Непредельные соединения, вступающие в реакцию с диенами, называются диенофилами.
Реакции полимеризации. Исключительно важной в практическом отношении является способность диеновых углеводородов вступать в реакцию полимеризации, в результате которой образуются каучукоподобные продукты.
Полимеризация протекает в 1,2- и 1,4-положения:
1 2 3 4 1 2 3 4
СН2=СН−СН=СН2 + СН2=СН−СН=СН2 + …. →
1 2 3 4 1 2 3 4
→ −СН2−СН=СН−СН2−СН2−СН=СН−СН2− (1,4-)
Реакция носит цепной характер, идет в присутствии инициаторов и катализаторов.
Алкины.
Непредельные соединения, содержащие в молекулу тройную связь, называют ацетиленовыми углеводородами или алкинами. Общая формула как у диеновых углеводородов, СnH2n-2.
Строение тройной связи было рассмотрено ранее – это комбинация двух π- и одной σ-связи. Длина тройной связи равна 0,120 нм, энергия образования 838,3 кДж/моль.
Номенклатура. В названиях алканов суффикс -ан заменяют на –ин. Главная цепь должна обязательно содержать тройную связь. Цепь нумеруют с того конца, к которому ближе расположена тройная связь.
Изомерия алкинов – изомерия положения тройной связи в цепи и углеродного скелета.
Способы получения.
1. Разложение карбида кальция водой.
СаС2 + 2Н2О → С2Н2 + Са(ОН)2
2. Дегидрогалогенирование дигалогенопроизводных, содержащих атомы галогена у одного или у соседних атомов углерода, действием спиртовой щелочи.
СН3 – СН2 – СНCl2 + 2KOH → CH3 – C ≡CH + 2KCl + 2H2O
3. Крекинг природного газа или нефти.
1500 °С
2СН4 → С2Н2 + 3Н2
4. Алкилирование ацетиленидов металлов.
СН3–I + Na–С≡СН → СН3−С≡СН + NaI
Физические свойства. (по Артеменко)
Химические свойства. Ацетиленовые углеводороды (алкины) способны вступать в реакции присоединения, замещения, окисления, полимеризации и конденсации.
Реакции присоединения. Реакции присоединения у алкинов протекают ступенчато: присоединяя одну молекулу реагента, тройная связь переходит в двойную, а присоединением второй молекулы – в одинарную. Имея две π-связи, алкины тем не менее медленнее вступают в реакции электрофильного присоединения, чем алкены. Это связано с устойчивостью промежуточных частиц: винильный катион, образующийся при присоединения электрофильной частицы в алкину, менее устойчив, чем карбокатион, образующийся при присоединении электрофила к алкену.
НС≡СН + А+ → НАС=СН+ (винил-катион)
Н2С=СН2 + А+ → Н2АС−СН2+ (карбокатион)
Энергия активации образования винил-катиона выше, чем для алкил-катиона, поэтому реакция электрофильного присоединения к алкинам протекает медленней, чем к алкенам.
Гидрирование. В присутствии катализаторов (Pt, Pd, Ni) происходит восстановление алкинов в алкены и алканы.
Н2 Н2
СН≡CН → CН2=СН2 → СН3 – СН3
Галогенирование ацетилена протекает с меньшей скоростью, чем реакция с этиленом. Br2 Br2
СН≡CН → CНBr=СНBr → СНBr2 – СНBr32
Гидрогалогенирование:
НСl НСl
СН≡CН → CН2=СНСl → СН3 – СНCl2
У алкенов эта реакция идет легче. Вторая молекула НСl присоединяется в соответствии с правилом Марковникова.
Присоединение воды (реакция Кучерова).
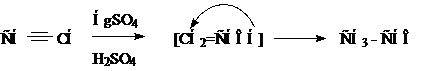
Присоединение органических кислот.
СН3 – СООН + СН≡CН → СН3 – СОО–СН=СН2
Сложный эфир – винилацетат применяют в качестве мономера для получения поливинилацетата.
Реакции полимеризации. Тримеризация ацетилена
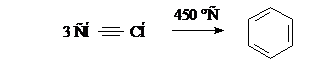
Реакции замещения. Атомы водорода в ацетилене обладают очень слабокислыми свойствами (как кислота в 1033 раз слабее НСl). В sp3-гибридном состоянии появляется небольшая кислотность за счет смещения электронной плотности.
δ+ δ− δ− δ+
Н→С≡C←Н
Такой поляризации достаточно, чтобы атомы водорода заменились на металл в щелочной среде.
СН≡CН + 2[Ag(NН3)2]OH → Ag−C≡C−Ag + 4NH3 + 2H2O
Лекция 7
Ароматические соединения.
Главным представителем ароматических углеводородов является бензол. Его строение изображают следующим образом:
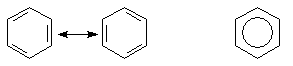
Формально бензол является циклогексатриеном-1,3,5. Однако в молекуле бензола все СС связи имеют одинаковую длину. Это связано с тем, что все 6 π-электронов равномерно распределены по молекуле бензола. В результате образуется единая устойчивая замкнутая электронная система. Этот эффект приводит к выигрышу в энергии который составляет 150,7 кДж/моль по сравнению с системой с тремя изолированными связями гипотетического циклогексатриена-1,3,5. Соединения типа бензола обладают совокупностью особых свойств. Соединения этого типа назвали ароматическими (бензол и его гомологи обладают сильным запахом).
Главной особенностью ароматических соединений является наличие делокализованной замкнутой системы π-электронов, подчиняющейся правилу Хюккеля: плоские циклические системы могут быть ароматическими, если они имеют сопряженную систему π-электронов и число этих электронов равно 4n+2, где n = 0, 1, 2, 3, и т.д..
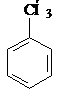
Изомерия и номенклатура. Общая формула гомологов бензола СnH2n-6. Ароматические углеводороды имеют изомеры, которые отличаются природой и положением заместителей в ароматическом ядре. Все шесть атомов водорода в бензоле совершенно одинаковы, поэтому существует только одно монометильное производное бензола – толуол.
При замещении двух атомов водорода образуется три изомера (орто-, мета- и пара-), которые отличаются положением заместителей.
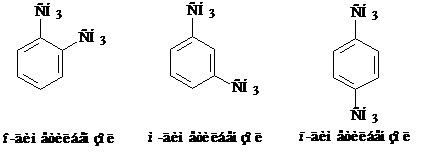
Вместо буквенного обозначения часто применяют цифровое: 1,2-диметил-бензол, 1,3-диметилбензол, 1,4-диметилбензол соответственно.
Изомеры могут отличаться и природой заместителей, которые бывают нормального и разветвленного строения.
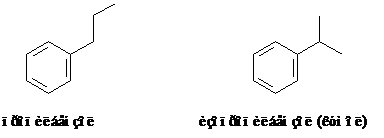
Кроме того, имея одинаковую брутто формулу, изомеры могут отличаться природой и положением заместителей. Например, углеводород состава С8Н10 имеет 4 изомера: один – этилбензол и три диметилбензола.
Для некоторых гомологов бензола используют тривиальные названия: винилбензол – стирол, метилбензол – толуол, диметилбензол – ксилол, и т.д.
Ароматические радикалы имеют общее название – арилы.
С6Н5−фенил, С6Н5−СН2− бензил, С6Н5−СН= бензаль,
С6Н5−С≡ бензо, С6Н4= фенилен.
Получение. Природные источники каменный уголь, коксовый газ, каменноугольная смола, нефть.
Синтез ароматических углеводородов.
1. Ароматизация парафинов. Например – каталитический реформинг С6 фракций нефти. Промежуточный продукт циклогексан, затем дегидрирование на Pt до бензола.
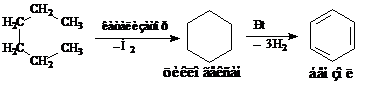
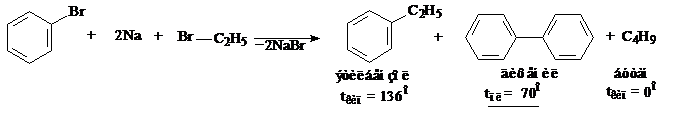
2. Реакция Вюрца – Фиттига. Эта реакция подобна реакции Вюрца, также образуется три продукта, но их легко разделить.
3. Синтез тримеризацией ацетилена (см. выше).
4. Реакция Фриделя – Крафтса.
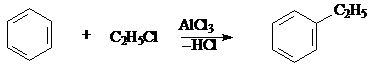
5. Восстановление по Клименсену.
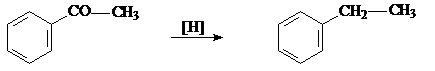
6. Синтез из солей ароматических кислот.
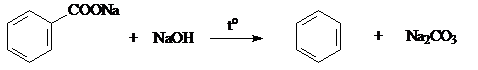
Химические свойства. В основном это реакции электрофильного замещения, а также некоторые реакции присоединения.
1.Реакции замещения. Это реакции электрофильного замещения (SE). К ним относятся реакции галогенирования, нитрования, сульфирования, алкилирования и ацилирования (реакции Фриделя-Крафтса) и некоторые другие.
а) Галогенирование
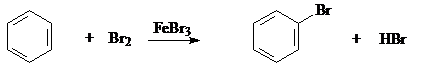
Бензол Бромбензол
б) Нитрование
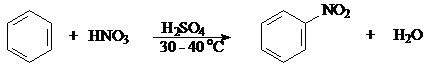
Бензол Нитробензол
в) Сульфирование
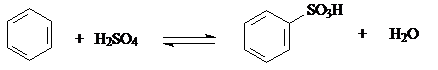
Бензол бензолсульфокислота (100%)
г) Алкилирование по Фриделю-Крафтсу.
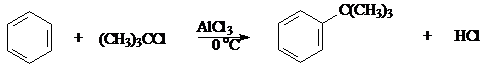
Бензол трет-бутилхлорид трет-бутилбензол (60%)
д) Ацилирование по Фриделю-Крафтсу
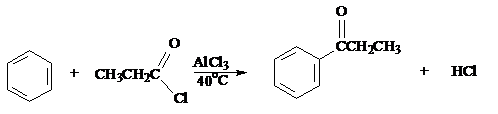
Бензол пропионилхлорид пропиофенон (90%)
В механизме электрофильного замещения есть общие закономерности. Реакции имеют ионный характер и протекают в несколько стадий.
Первой стадией электрофильного замещения является образование π-комплекса в результате взаимодействия π-электронной системы бензольного кольца с заряженной электрофильной частицей или нейтральным электрофильным реагентом (SO3).
X―Y → X+ + Y–

π- комплекс 1
Вторая стадия состоит в переходе π-комплекса в σ-комплекс (бензониевый ион). Это происходит в результате выведения из сопряженной системы шести электронов двух электронов для образования новой ковалентной связи С―Х. Оставшиеся четыре π-электрона распределяются между пятью углеродными атомами бензольного кольца.
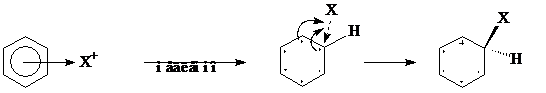
Образовавшийся σ-комплекс можно изобразить иначе, в виде пентадиенильного катиона. Граничные (крайние) структуры могут быть представлены следующим образом:
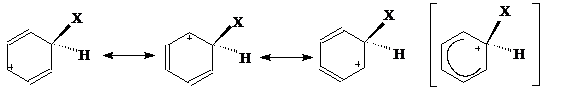
σ- комплекс (бензониевый ион)
σ-Комплекс – промежуточная частица представляет собой неустойчивый не ароматический карбокатион. Шесть его углеродных атомов находятся в разных валентных состояниях: один – насыщенный в первом валентном состоянии (sp3-гибридизация), а пять других - в обычном для бензола втором валентном состоянии (sp2-гибридизация) Атом Х (или группа) и водород при насыщенном углеродном атоме расположены в плоскости перпендикулярной плоскости бензольного кольца.
Процесс образования карбокатиона подобен образованию карбокатиона в случае электрофильного присоединения НХ к алкенам, но стабилизация карбокатионов протекает различным образом. В случае алкенов – за счет присоединения аниона, в случае бензониевого иона – за счет выброса протона с восстановлением ароматичности.
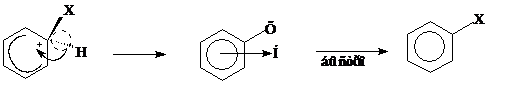
σ- комплекс π- комплекс 2 замещенный бензол
Механизм электрофильного замещения бензола иллюстрируется энергетической диаграммой реакции
Лекция 8.
Классификация заместителей. Понятие об ориентирующем влиянии заместителей
Если в бензольном кольце находится хотя бы один заместитель
то в нем неизбежно нарушение равномерного распределения π–электронной плотности. В результате молекула бензола становится частично поляризованной, что определяет вхождение атакующего реагента в то или иное положение кольца.
Толуол, подобно бензолу, подвергается электрофильному ароматическому замещению, например сульфированию. Хотя возможно образование трех моносульфопродуктов, в действительности в ощутимых количествах получается только два изомера – орто и пара
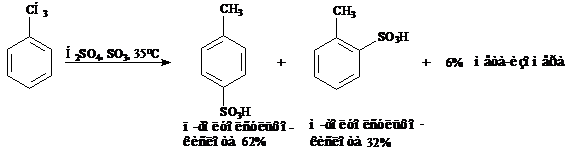
Изучение нитрования, галогенирования и реакции алкилирования по Фриделю-Крафтсу толуола дает аналогичные результаты. Метильная группа делает кольцо более реакционноспособным, чем незамещенный бензол направляет атаку в орто- и пара-положения кольца.
Совершенно иначе ведет себя нитробензол, который реагирует медленнее, чем бензол, и дает главным образом мета-изомер.
Подобно метильной или нитрогруппе, любая группа, связанная с бензольным кольцом, влияет на реакционную способность кольца и определяет ориентацию замещения. Когда электрофильный реагент атакует ароматическое кольцо, именно природа группы, уже имеющейся в кольце, определяет, насколько легко происходит атака и ее направление.
Группа, под влияние которой кольцо становится более активным, чем кольцо бензола, называется активирующей группой. Группа, делающая его менее активным, чем кольцо бензола, называется дезактивирующей группой.
Группа, направляющая атаку преимущественно в орто- и пара-положения, называется орто- и пара-ориентантом. Группа, которая направляет атаку преимущественно в мета-положение, называется мета-ориентантом.
Определение ориентации. В принципе очень просто выяснить характер ориентирующего влияния группы: для этого надо ввести в реакцию замещения вещество, содержащее в качестве заместителя у бензольного кольца эту группу, и проанализировать образующийся продукт на содержание трех изомеров. Идентификация каждого из изомеров как орто- мета- или пара- обычно производится путем сравнения с заведомым образцом этого изомера, полученным каким либо другим методом из соединения, структура которого известна. Именно таким путем было определено к какому из двух классов: орто-, пара- ориентанты или мета-ориентанты относится та или иная группа.
Из пяти возможных для атаки положений три (60%) находятся в орто- и пара-положениях к заместителю, а два (40%) – в мета-положении к заместителю. Если реакция замещения протекала бы не избирательно, то следовало бы ожидать, что продукт будет состоять на 60% из орто- и пара-изомеров, и на 40% из мета-изомера.
В таблице 1 приведены данные по ориентации при реакции нитрования ряда замещенных бензолов.
Таблица 1
Ориентация при нитровании С6Н5Y
| ―Y | орто | пара | орто+пара | мета |
| ―ОН | 50-55 | 45-50 | следы | |
| ―NHCOCH3 | ||||
| ―CH3 | ||||
| ―F | следы | |||
| ―Cl | следы | |||
| ―Br | ||||
| ―I | ||||
| ―NO2 | 6,4 | 0,3 | 6,7 | 93,3 |
| ―N+(CH3)2 | ||||
| ―CN | ||||
| ―COOH | ||||
| ―SO3H | ||||
| ―CHO |
Как видно из таблицы, семь групп направляют нитрование на 96-100% в орто- и пара-полжения, а шесть других – на 72-94% в мета-положение.
Определенная группа проявляет одинаковое ориентирующее действие – преимущественно в орто и пара или преимущественно мета – независимо от природы электрофильного реагенты.
Определение относительной реакционной способности. Реакционную способность бензола и замещенных бензолов сравнивают одним из следующих способов.
Во-первых, можно определить время, необходимое для протекания реакции в идентичных условиях. Например, толуол реагирует с дымящей серной кислотой в 10-20 раз быстрее, чем бензол. Таким образом, толуол более активен, чем бензол, и следовательно, СН3-группа является активирующей группой.
Во-вторых, можно устанавливать степень жесткости условий, требуемых для протекания сравниваемых реакций за один и тот же период времени. Например, бензол нитруется менее чем за час при 60 оС смесью концентрированных серной и азотной кислот. Сравнимая степень нитрования нитробензола требует использования температуры 90 оС и смеси дымящей азотной и концентрированной серной кислот. Очевидно, нитробензол менее активен, чем бензол, и нитрогруппа NO2 является дезактивирующей группой.
В-третьих, для точных количественных сравнений необходимо в идентичных условиях проводить конкурентные реакции, в которых сравниваемые соединения вводят в реакция с недостаточным количеством реагента. Так, например,если эквимолярное количество бензола и толуола обрабатывают небольшим количеством азотной кислоты, то образуется примерно в 25 раз больше нитротолуолов, чем нитробензола. Это свидетельствует о том, что толуол в 25 раз активнее бензола.
Классификация заместителей. Почти все группы относятся к одному из двух классов: активирующие и орто-, пара-ориентирующие или дезактивирующие и мета-ориентирующие. Галогены составляют отдельную группу заместителей: дезактивирующих, но орто-, пара-ориентирующих.
К орто-, пара-ориентанттам (заместителям первого рода) относят все электронодонорные заместители и галогены:
ОН, О–, OR, OC6H5, NH2, NHR, NR2, NHCOCH3, NHCOR, CH3 и другие алкилы, СH2Cl.
Причина орто-, пара-ориентирующего влияния перечисленных заместителей заключается в том, что за счет положительного мезомерного эффекта они в большей степени подают электронную плотность в орто- и пара-положения и в большей степени стабилизируют σ-комплексы, образующиеся при вступлении электрофильного агента в эти положения.
Заместители ОН, О–, OR, OC6H5, NH2, NHR, NR2, NHCOCH3, NHCOR, характеризуются большим положительным эффектом сопряжения по сравнению отрицательным индуктивным эффектом.
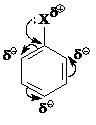
+M > −I
Эти заместители являются активирующими. Электрофильное замещение в соответствующих монозамещенных бензолах протекает быстрее, чем в бензоле.
К активирующим заместителям относят также метильную группу и другие алкильные группы, для которых характерны электронодонорные эффекты: сверхсопряжение и индуктивный эффект.
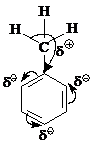
+I , +M
Галогены также являются орто-, пара-ориентантами, но при этом дезактивируют электрофильное замещение, поскольку для них характерно большее значение отрицательного индуктивного эффекта по сравнению с положительным мезомерным эффектом (–I > +M): оттягивая электрон
| <== предыдущая страница | | | следующая страница ==> |
| | | Пояснительная записка. Данные методические рекомендации для организации самостоятельной работы по литературе предназначены для студентов 1 курса |
Дата добавления: 2014-10-14; просмотров: 1847; Нарушение авторских прав

Мы поможем в написании ваших работ!