Главная страница Случайная лекция

Мы поможем в написании ваших работ!
Порталы:
БиологияВойнаГеографияИнформатикаИскусствоИсторияКультураЛингвистикаМатематикаМедицинаОхрана трудаПолитикаПравоПсихологияРелигияТехникаФизикаФилософияЭкономика

Мы поможем в написании ваших работ!
КАРБОНОВЫЕ КИСЛОТЫ
В состав органических карбоновых кислот в качестве функциональной входит карбоксильная группа
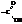
В зависимости от числа этих групп различают одно-, двух-, трёх-… многоосновные кислоты. В зависимости от типа присоединенного к карбоксилу радикала они обычно подразделяются на алифатические (насыщенные и ненасыщенные), алициклические, ароматические и т.д.
5.1 Алифатические карбоновые кислоты
5.1.1 Одноосновные насыщенные кислоты
Гомологический ряд этих кислот начинается муравьиной кислотой
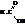
Изомерия кислот этого ряда определяется строением углеводородного радикала при карбоксильной группе и начинается с четвёртого члена ряда. Тривиальные названия карбоновых кислот привились очень прочно и широко используются наряду с названиями по номенклатуре ИЮПАК. Названия кислот по этой номенклатуре производятся от названия соответствующего углеводорода с добавлением суффикса -овая кислота. Иногда эти кислоты называют как производные уксусной кислоты.

| муравьиная, метановая | |
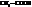
| уксусная, этановая | |
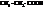
| пропионовая, пропановая | |

| масляная, бутановая | |
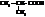
| изомасляная, 2-метилпропановая | |
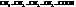
| валериановая, пентановая | |
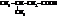
| изовалериановая, 3-метилбутановая | |
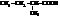
| метилэтилуксусная, 2-метилбутановая | |
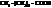
| капроновая, гексановая | |
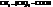
| энантовая, гептановая | |

| каприловая, октановая | |
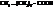
| пеларгоновая, нонановая | |
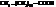
| пальмитиновая, гексадекановая | |
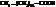
| стеариновая, октадекановая и т.д. |
Способы получения
– Способы, основанные на окислении:
а) окисление первичных спиртов (см. тему «Гидроксисоединения»);
б) окисление альдегидов (см. тему «Карбонильные соединения»);
в) окисление предельных углеводородов кислородом воздуха на катализаторе. При окислении синтина (см. тему «Алканы») этот способ даёт смесь кислот с разной длиной цепи и используется в промышленности при производстве моющих средств.
– Гидролиз нитрилов, которые получают действием цианида калия на галогеналкилы
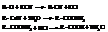
– Гидролиз геминальных тригалогеналканов 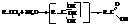
– Синтез через магнийорганические соединения
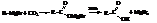
– Оксосинтез – промышленный метод получения карбоновых кислот. Он заключается в действии на олефины оксида углерода (II) и водяного пара при 300…400 °С в присутствии катализаторов (например, Ni(CO)4)
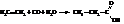
Физические свойства
Муравьиная, уксусная и пропионовая кислоты легкоподвижные бесцветные жидкости с характерным острым запахом. С водой смешиваются во всех отношениях. Кислоты С4 – С9 – маслянистые жидкости. Растворимость их в воде сильно уменьшается с ростом молекулярной массы. Кислоты С10 и выше – твёрдые кристаллические вещества, нерастворимые в воде.
Температуры кипения их растут по мере увеличения молекулярной массы. Сравнительно со спиртами, имеющими то же число углеродных атомов, кислоты кипят выше. Например, этиловый спирт кипит при
78,3 °С, а уксусная кислота – при 118,5 °С. Это объясняется большей ассоциацией молекул кислоты и образованием ею более прочных ассоциатов. Физические измерения указывают в основном на димеризацию за счёт водородных связей.
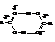
Кислоты с чётным числом углеродных атомов плавятся при более высокой температуре, чем кислоты с нечётным числом.
Химические свойства
– Карбоновые кислоты – это протонные кислоты. При диссоци-ации они выделяют протон
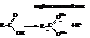
Кислотные свойства объясняются перераспределением электронной плотности в карбоксильной группе в сторону кислорода.
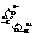
Вследствие этого связь О – Н ослабляется и сравнительно легко разрывается. Величина константы диссоциации фактически зависит от величины дробного положительного заряда на карбоксильном углероде. Чем он выше, тем сильнее кислота. Степень диссоциации или, другими словами, сила кислот зависит также от величины и характера радикала, соединённого с карбоксильной группой. Алкильные радикалы посредством индукционного эффекта повышают электронную плотность в карбоксиле (понижают положительный заряд на карбоксильном углероде), тем самым, понижая силу кислоты.

С увеличением алкильного радикала константы диссоциации карбоновых кислот несколько уменьшается. Так, константа диссоциации муравьиной кислоты Н–СООН 2,17·10-4, уксусной СН3–СООН 1,76·10-5 и капроновой СН3–СН2–СН2–СН2–СН2–СООН 1,31·10-5.
Введение в радикал электрофильной, электронооттягивающей группы, напротив, увеличивает силу кислоты. К примеру, константа диссоциации хлоруксусной кислоты 1,40·10-3.
– Как всякие кислоты, карбоновые кислоты способны к образованию солей с металлами, их оксидами и гидрооксидами
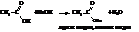
Пиролизом кальциевых солей получают альдегиды и кетоны, пиролизом натриевых в присутствии NаОН – углеводороды.
– При действии на кислоты галогенидов фосфора или тионилгалогенида образуются галогенангидриды кислот
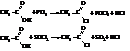
Галогенангидриды кислот называют по кислоте и галогену (хлорангидрид уксусной кислоты), а также по кислотному радикалу ацилу
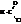
| хлористый формил, хлористый метаноил |
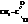
| хлористый ацетил, хлористый этаноил |
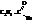
| Бромистый пропионил, бромистый пропаноил |
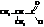
| хлористый изобутирил, хлористый 2-метилпропаноил |
В галоидангидридах кислот галоид чрезвычайно подвижен и легко вступает в реакции обмена. Причиной подвижности галоида является большой положительный заряд на углероде (δ+), который связан сразу с двумя электронооттягивающими атомами – кислородом и галогеном.
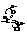
Этот углерод легко подвергается атаке нуклеофильных реагентов.
В реакциях галогенангидридов с соединениями, содержащими металлы или активный атом водорода, происходит замена атомов металла или водорода на ацил. Реакция называется ацилированием. Например, в случае уксусной кислоты – это ацетилирование.
Ацилированием получают целый ряд производных кислот: соли, ангидриды, сложные эфиры, амиды, нитрилы и др. Например:
а) гидролиз хлорангидрида или ацетилирование воды

б) алкоголиз хлорангидрида или ацетилирование спиртов
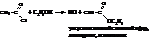
в) аммонолиз хлорангидрида или ацетилирование аммиака
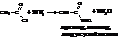
г)

– В результате отщепления воды от двух молекул кислоты также образуются ангидриды кислот
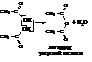
или ангидрид этановой кислоты
Таким путём ангидриды получаются лишь в очень жёстких условиях: или в присутствии сильных водоотнимающих средств (Р2О5), или в присутствии катализаторов (Al2O3) при высоких температурах. Обычно их синтезируют действием галогенангидридов на соли кислот
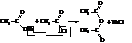
Ангидрид могут образовывать и две различные кислоты.
Ангидриды кислот – вещества весьма активные:
с водой они дают соответствующие кислоты
 ,
,
со спиртами и аммиаком образуют смеси кислот со сложными эфирами и амидами соответственно.
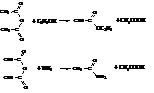
Ангидриды кислот являются хорошими ацилирующими средствами. Однако половина молекулы ангидрида не ацилирует, а выделяется в виде кислоты.
– Амиды кислот. Их обычно получают через галогенангидриды и ангидриды кислот (см. выше). В промышленности амиды синтезируют действием аммиака на кислоты
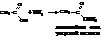
При сухой перегонке аммонийная соль выделяет воду с образованием амида
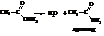
Ацетамид по номенклатуре ИЮПАК будет называться этанамидом.
При ацилировании аминов получаются алкил- и диалкиламиды, имеющие промышленное значение.
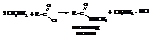
– Сложные эфиры можно получать прямым взаимодействием кислоты со спиртом, т.е. реакцией этерификации кислот спиртами в присутствии кислого катализатора
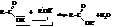
Эта реакция давно интересовала химиков. В 1862 году Бертло установил, что реакция обратима и равновесие наступает, когда прореагирует примерно по 2/3 моля исходных веществ. В конце
ХIХ века Н. А. и Н. Б. Меншуткины обнаружили, что скорость реакции этерификации зависит от строения кислоты и спирта. Было установлено, что скорость этерификации падает при увеличении числа и объёма радикалов при α-углеродном атоме по отношению к карбоксилу. Этот факт, очевидно, объясняется экранированием углеродного атома карбоксила замещающими радикалами.
С помощью меченых атомов (изотоп кислорода О18) было показано, что вода при этерификации образуется за счёт гидроксила кислоты и водорода спирта
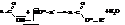
Названия сложных эфиров по тривиальной номенклатуре строятся по следующей схеме: название радикала спирта плюс название кислотного радикала, в котором окончание -ил заменено на -ат. По номенклатуре ИЮПАК: название радикала спирта плюс название кислоты, в котором окончание -овая заменено на -оат.
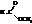
| метилформиат, метилметаноат |
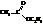
| этилацетат, этилэтаноат |

| бутилпропионат, бутилпропаноат |
Как показано выше, сложные эфиры можно также получать взаимодействием спиртов с ангидридами и галогенангидридами кислот.
В реакции со спиртами сложные эфиры способны обмениваться с ними радикалами. Эта реакция называется переэтерификацией.
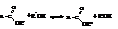
– Галогензамещенные кислоты
Хотя в карбоксильной группе наблюдается значительное смещение электронной плотности к кислороду, углеродный атом карбоксила обладает не столь большим положительным зарядом, как углерод карбонила в кетонах и альдегидах. Это обусловлено частичной нейтрализацией электронной ненасыщенности углерода смещением электронов от гидроксила
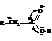
И всё же карбоксил оказывает влияние на соединённый с ним углеводородный радикал, оттягивая на себя электронную плотность и повышая активность водородных атомов, прежде всего у α-углерода.
Прямое хлорирование и бромирование карбоновых кислот происходит на свету с вступлением галогенов в α-положение.

Обычным же способом синтеза α-галогензамещённых кислот является метод Гелль-Фольганд-Зелинского, заключающийся в действии на карбоновую кислоту молекулярного хлора или брома в присутствии фосфора
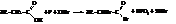
Другой метод синтеза, приводящий к β-галогензамещенным кислотам, заключается в гидрохлорировании α,β-ненасыщенных карбоновых кислот
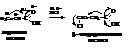
Присоединение НCl протекает вопреки правилу Марковникова из-за смещения электронной плотности по системе сопряжения молекулы к кислороду и в соответствии с зарядами атомов.
Действие галогеноводородных кислот на гидроксикислоты также приводит к желаемому результату
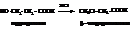
Замещение α-водорода галогеном значительно повышает силу кислоты. Галоген сильно оттягивает электронную пару от α-углерода, обнажая его положительный заряд. Он, в свою очередь, через углерод карбоксила притягивает к себе электроны гидроксильной группы. Таким образом, посредством индукционного эффекта, протон гидроксила активируется. Подобная картина, правда, менее выражено, наблюдается и при удалении галогена от карбоксила.
При замене водорода в радикале кислоты на I, Br, Cl, F, а также увеличении числа атомов галогена, последовательно увеличивается сила и константы диссоциации кислот (К).
Например:
| К·105 | К·105 | ||

| 1,75 | 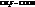
| |
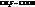
| 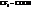
| ||
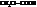
| 
| ||
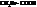
| 
| ||
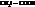
|
Галогенкислоты благодаря большой активности нашли широкое применение.
Отдельные представители
Муравьиная кислота– жидкость с tкип = 100,8 °C и резким запахом. При попадании на кожу вызывает ожоги.
В промышленности её получают из оксида углерода и едкого натра
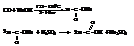
Муравьиная кислота занимает особое место в ряду кислот. Наряду с карбоксилом в ней может быть выделена и альдегидная группа.
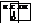
Поэтому, обладая всеми свойствами кислот, она вступает в реакции окисления: серебряного зеркала, с фелинговой жидкостью, – т.е. в типично альдегидные реакции.
Муравьиная кислота используется для дезинфекции в медицине и промышленности, при крашении тканей, в ряде синтезов.
Уксусная кислота
Безводная уксусная кислота при температуре +16,6 °С застывает и называется поэтому ледяной. tкип.= 118,5 °С.
Получают её гидратацией ацетилена, окислением этилового спирта и предельных углеводородов, уксуснокислым брожением сахаристых веществ и другими методами.
В химическом отношении – это обычная карбоновая кислота.
Применяется как прекрасный растворитель, в кожевенном производстве, лакокрасочной, пищевой, химической и других отраслях промышленности. Причём расходуется в больших количествах.
Высшие жирные кислоты
Наиболее известны пальмитиновая С15Н31СООН и стеариновая С17Н35СООН кислоты. Эти кислоты имеют молекулы с нормальной цепочкой углеродных атомов. Их получают при омылении жиров и каталитическим окислением парафинов.
Соли этих кислот – мыла. Натриевые и калиевые соли этих кислот хорошо растворимы в воде и хорошо «мылятся». Соли магния, кальция, бария и др. (соли жёсткости) плохо растворяются в воде. Поэтому в жёсткой воде обычные мыла переходят в нерастворимое состояние и не «мылятся».
Для изготовления свечей используют твердую смесь высших жирных кислот – стеарин.
5.1.2 Одноосновные ненасыщенные кислоты
К ряду ненасыщенных карбоновых кислот можно отнести следующие:

| акриловая, пропеновая | |
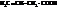
| винилуксусная, бутен-3-овая | |

| кротоновая, бутен-2-овая | |

| метакриловая, 2-метилпропеновая | |

| ||
| олеиновая, октадецен-9-овая | ||

| ||
| линолевая, октадекадиен-9,12-овая | ||

| ||
| линолевая, октадекатриен-9,12,15- овая | ||

| ||
| арахидоновая, эйкозатетраен-5,8,10,13-овая | ||
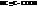
| Про пропиоловая, пропиновая |
Необходимо отметить, что кислоты типа
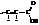
с сопряжёнными двойными связями (карбонильной и этиленовой) обычно называют a,b-ненасыщенными кислотами. Они резко отличаются по способам получения и свойствам от кислот с изолированными двойными связями.
К последним относятся олеиновая, линолевая, линоленовая, арахидоновая кислоты, остатки которых входят в состав молекул жиров.
К α,β–ненасыщенным кислотам, относятся акриловая и метакри-ловая кислоты.
Акриловая кислота – жидкость с резким запахом, tкип.= 140 °С.
В промышленности получают из этилена или ацетилена
 В промышленности в основном используются производные акриловой кислоты, прежде всего, сложные эфиры кислоты. Например, метиловый эфир – метилакрилат – легко полимеризуется и используется, поэтому в производстве органического стекла и других полимеров.
В промышленности в основном используются производные акриловой кислоты, прежде всего, сложные эфиры кислоты. Например, метиловый эфир – метилакрилат – легко полимеризуется и используется, поэтому в производстве органического стекла и других полимеров.
Акриловая кислота и соответствующий ей альдегид, акролеин, образующиеся в небольших количествах при длительном нагревании растительных масел (приготовление олифы, чипсов), оказывает негативное воздействие на живой организм.
Кротоновая кислота
Эта кислота существует в форме двух геометрических изомеров: цис-изомер называют изокротоновой, транс-изомер – кротоновой кислотой.
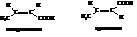
Кротоновая кислота является энергетически более устойчивой формой по сравнению с изокротоновой.
Из высших ненасыщенных кислот наибольшее значение имеет олеиновая кислота. Она также представлена цис- и транс-изомерами. Причём транс-форма, называемая эландиновой кислотой, на
32,2 кДж∙моль-1 устойчивее цис-олеиновой кислоты.
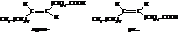
Щелочные соли олеиновой кислоты являются мылами и используются в технике, остатки цис-олеиновой кислоты входят в состав молекул жиров.
5.1.3 Двухосновные насыщенные кислоты
Примерами предельных двухосновных кислот могут служить:
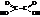
| щавелевая, этандиовая (дикарбоновая) |

| малоновая, пропандиовая |
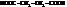
| янтарная, бутандиовая |
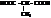
| метилмалоновая, метилпропандиовая |

| глутаровая, пентандиовая |
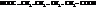
| адипиновая, гександиовая и т.д. |
Кислоты этого ряда являются кристаллическими веществами, хорошо растворимыми в воде. Также как и в ряду одноосновных кислот, температуры плавления двухосновных кислот с чётным числом углеродных атомов выше температур плавления. соседних гомологов с нечётным числом углеродов.
Константа диссоциации первого карбоксила из-за мощного взаимного влияния карбоксильных групп выше, чем у одноосновных кислот.
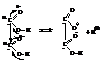
В то же время константа диссоциации второго карбоксила ниже, чем у монокарбоновых кислот, вследствие большого (+2) отрицательного заряда иона, удерживающего протон.
Двухосновные кислоты имеют те же химические свойства, что и монокарбоновые, только в реакции могут вступать два карбоксила. Вместе с тем присутствие в молекуле сразу двух карбоксилов обуславливает и некоторые особенности в их химическом поведении.
Например, щавелевая и малоновая кислоты при нагревании выше температуры плавления разлагаются с отщеплением в виде диоксида углерода одного карбоксила
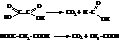
Янтарная и глутаровая кислоты способны к внутримолекулярному отщеплению воды при нагревании. В результате получаются внутренние ангидриды
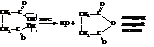
При пиролизе кальциевых солей адипиновой кислоты и некоторых её гомологов происходит образование циклических кетонов.
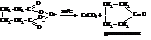
Щавелевая кислота весьма распространена в природе: кислая калиевая соль её НООС – СООК находится в щавеле, кислице, щавеле-во-кислый или оксалат кальция – во многих растениях.
В технике щавелевую кислоту получают окислением древесных опилок кислородом воздуха при нагревании их с расплавом КОН или при быстром нагревании до температуры 400 °С калиевой или натриевой соли муравьиной кислоты.

Щавелевая кислота легко окисляется до СО2 и Н2О и при быстром нагревании разлагается
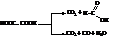
Она является очень сильной кислотой. Константа диссоциации её примерно в 2000 раз выше, чем у уксусной кислоты.
Щавелевая кислота используется в ситцепечатании, кожевенной и пищевой промышленности и во многих других отраслях народного хозяйства.
Малоновая кислота
Вследствие влияния двух карбоксилов водороды метиленовой группы малоновой кислоты активируются и легко замещаются.
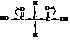
В лабораториях очень широко практикуется синтез одно- и двухосновных кислот через эфиры малоновой кислоты.
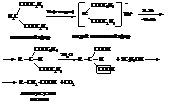
Янтарная кислота используется для получения некоторых пластмасс и в качестве пластификатора.
Адипиновая кислота используется в производстве полиамидных смол и волокна найлон.
5.1.4 Двухосновные ненасыщенные кислоты
Наиболее простыми представителями этого ряда кислот являются фумаровая и малеиновая кислоты. Они имеют одинаковую структурную формулу и являются геометрическими изомерами:
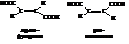
Обе кислоты получают из яблочной кислоты. Осторожное нагревание даёт фумаровую, сильное – малеиновую кислоту
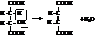
Одинаковость структурной формулы обеих кислот подтверждается тем, что при гидрировании они дают одну кислоту – янтарную
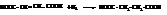
Различия в пространственном строении малеиновой и фумаровой кислот сильно отражается на их свойствах.
Фумаровая кислота плохо растворима в воде, возгоняется без плавления при температуре 200 °С. Малеиновая кислота очень легко растворима в воде и плавится при 130 °С, она является более сильной кислотой, чем фумаровая.
При нагревании малеиновая кислота легко отщепляет воду с образованием малеинового ангидрида
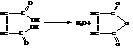
Малеиновый ангидрид получается в технике при неполном окислении многих органических соединений (бензол, бутилены, циклогексан и др.). Он является хорошим диенофилом и применяется для обнаружения сопряженных диенов. Используется также в производстве стеклопластиков и для получения сополимеров со стиролом, акриловыми и метакриловыми эфирами. Такое применение его обусловлено наличием в молекуле активированной двойной связи С = С.
Фумаровая кислота своего ангидрида не образует и лишь при температуре 300 °С отщепляет воду с образованием… малеинового ангидрида.
С энергетической точки зрения фумаровая кислота стабильней малеиновой кислоты на 29,2 кДж∙моль-1.
5.2 Ароматические карбоновые кислоты
В зависимости от числа карбоксильных групп, – СООН, связанных с ароматическим ядром, ароматические карбоновые кислоты могут быть одно-, двух- и многоосновными. Они обычно носят тривиальные названия. Например:
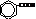
| бензойная кислота |
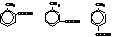
| о-, м- и п-толуиловые кислоты |
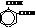
| о-фталевая |
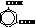
| м-фталевая или изофталевая |

| п-фталевая или терефталевая |
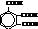
| 1,2,3-бензолтрикарбо-новая кислота или гемимеллитовая |
Способы получения
Ароматические карбоновые кислоты могут быть получены обычными методами получения карбоновых кислот, например, гидролизом геминальных тригалогенпроизводных с галогенами в боковой цепи, гидролизом ароматических нитрилов, через магнийорганические соединения и т.д.. Однако преимущественно их получают способами, основанными на окислении.
– Окисление ароматических углеводородов. При энергичном окислении боковые цепи гомологов бензола «отгорают» и образуют соответствующие ароматические кислоты. Окислителями служат хромовая кислота и щелочной раствор перманганата калия.
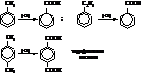
– Окисление ароматических кетонов гипохлоритом натрия
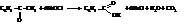
Физические свойства
Ароматические кислоты – кристаллические вещества, плохо растворимые в воде и хорошо – в полярных органических растворителях.
Химические свойства
– Константы диссоциации ароматических кислот несколько выше, чем у кислот жирного ряда (.бензойная кислота – 6,6·10-5,.уксусная кислота – 1,76·10-5). Это связано с большей электрофильностью бензольного кольца (смещение электронной плотности к сильно ненасыщенной группировке).
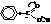
– Для карбоксильной группы в ароматических кислотах характерны те же реакции, что и в алифатическом ряду: это реакции образования солей со щелочами, образования ангидридов и галогенангидридов, реакции этерификации, ацилирования и т.д.
Например, при нагревании смеси ароматической кислоты и спирта в присутствии серной кислоты образуется сложный эфир.

Если заместителей в орто-положении нет, то этерификация идёт также легко, как и в случае алифатических кислот. Однако, если одно из орто-положений замещено, скорость реакции резко падает, если же замещены оба орто-положения – реакция вовсе не идет.
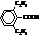
Этот факт указывает на пространственные затруднения: орто-заместители настолько сильно экранируют углерод кислотной группы, что затрудняют или делают невозможной атаку внешнего реагента на него.
В реакциях замещения в ароматическом ядре карбоксильная группа выступает как ориентант второго рода (электроноакцепторная группа) и направляет электрофильную атаку в мета-положение, затрудняя реакцию.
Бензойная кислота
Бензойная кислота содержится в некоторых природных смолах. Получают её почти исключительно окислением толуола. Она применяется в производстве красителей и лекарственных веществ. Большое промышленное значение имеет хлористый бензоил – хлорангидрид бензойной кислоты,
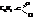
Получают его неполным гидролизом бензотрихлорида, который готовят хлорированием толуола на свету.
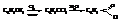
Хлористый бензоил находит применение как ацилирующее средство.
Из ароматических кислот с карбоксилом в боковой цепи наиболее известна фенилуксусная кислота, которую получают через её нитрил.
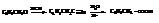
Используют в парфюмерии и медицине.
Дикарбоновые ароматические кислоты
Они представлены тремя изомерами
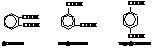
Используются, в основном, фталевая и терефталевая кислоты. Фталевая кислота получается окислением о-ксилола (о-диметилбензола) или нафталина. При нагревании она даёт фталевый ангидрид.
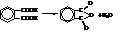
Фталевая кислота используется, главным образом, в виде фталевого ангидрида. Он идёт на получение эфиров фталевой кислоты. Диметилфталат является хорошим репелленом (отпугивает комаров, гнус). Конденсацией фталевого ангидрида и глицерина получают глифталевые смолы, применяющиеся в качестве прочных покровных материалов, плёнок, лаков. Фталевый ангидрид идёт на производство красителей.
Терефталевую кислоту получают окислением п-ксилола. Она применяется для получения полиэтилентерефталата, из которого формуют синтетическое полиэфирное волокно лавсан. Этерификация кислоты этиленгликолем протекает как реакция поликонденсации с образованием полимера и выделением воды:
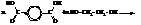
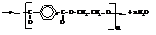
ЖИРЫ
Компоненты смеси органических веществ, экстрагируемых из тканей животных или растений неполярными растворителями (диэтиловый эфир, хлороформ, бензол, алканы), называют липидами. К липидам относят следующие совершенно различные по строению вещества: карбоновые кислоты, триглицериды или жиры, фосфолипиды и гликолипиды, воски, терпены, стероиды. Это соединения нерастворимые в воде и хорошо растворимые в органических растворителях.
Основная часть эфирной вытяжки – это собственно жиры или глицериды: сложные эфиры трёхатомного спирта глицерина и высших жирных кислот.
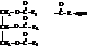
Жиры являются необходимой и весьма ценной составной частью пищи. Они обладают высокой калорийностью и в значительной степени снабжают организм энергией. При окислении 1г жира выделяется ~40 кДж энергии (1г углеводов ~17 кДж; 1г белка ~23 кДж). Жиры в организме вследствие их энергетической ценности служат резервным питательным веществом. После приема в пищу жиров долго сохраняется ощущение сытости. Суточный рацион человека 60…70 г жиров. В природных жирах в качестве примесей содержатся и другие полезные вещества, в том числе витамины А, D, Е. Жиры служат также теплоизоляционным материалом, затрудняющим охлаждение организма.
В кишечнике под влиянием фермента липазы жиры гидролизуются до глицерина и органических кислот. Продукты гидролиза всасываются стенками кишечника и синтезируются новые жиры. (В организмах животных и растений входящие в состав жиров высшие предельные жирные кислоты синтезируются из уксусной кислоты, глицерин - из глюкозы). Кислоты с несколькими двойными связями (линолевая, линоленовая) синтезируются только растениями и поэтому являются незаменимыми компонентами пищи. В организмах животных они необходимы как исходные материалы в синтезе простагландинов, недостаток которых вызывает замедление роста, поражение кожи, нарушение функции почек, органов размножения.
Жиры широко используются в технических целях для изготовления мыл, олифы, линолеума, клеенки, смазочных материалов, а также в медицине и парфюмерии.
Физические свойства
Жиры легче воды и нерастворимы в ней. Хорошо растворимы в органических растворителях, например, в бензине, диэтиловом эфире, хлороформе, ацетоне и т.д. Температура кипения жиров не может быть определена, поскольку при нагревании до 250 °С они разрушаются с образованием из глицерина при его дегидратации сильно раздражающего слизистые оболочки глаз альдегида - акролеина (пропеналя).
Для жиров прослеживается довольно четкая связь химического строения и их консистенции. Жиры, в которых преобладают остатки насыщенных кислот – твёрдые (говяжий, бараний и свиной жиры). Если в жире преобладают остатки ненасыщенных кислот, он имеет жидкую консистенцию. Жидкие растительные жиры называется маслами (подсолнечное, льняное, оливковое и т.д. масла). Организмы морских животных и рыбы содержат жидкие животные жиры. В молекулы жиров мазеобразной (полутвёрдой) консистенции входят одновременно остатки насыщенных и ненасыщенных жирных кислот (молочный жир).
Изомерия и номенклатура
Как уже отмечалось, жиры – это сложные эфиры глицерина и высших жирных кислот. В жирах найдено до 200 различных жирных кислот с содержанием обычно четного числа атомов углерода
от 4 до 26. Наиболее часто встречаются кислоты с 16 и 18 атомами углерода в цепи. В состав молекул жиров могут входить остатки одинаковых и разных кислот (ацилы).
Природные триглицериды обычно содержат остатки двух или трех различных кислот. В зависимости от того одинаковые или разные остатки кислот (ацилы) входят в состав молекул жира они делятся на простые и смешанные.
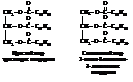
Структурная изомерия характерна прежде всего для смешанных жиров. Так, для показанного выше смешанного триглицерида возможны три структурных изомера с различным расположением ацильных остатков при углеродах глицерина. Теоретически для жиров, в состав которых входят остатки ненасыщенных высших жирных кислот, возможна геометрическая изомерия у двойных связей и изомерия, обусловленная различным положением двойных связей. Однако, хотя остатки ненасыщенных жирных кислот в природных жирах встречаются чаще, двойная связь в них обычно располагается между углеродами С9-С10, причем этиленовая группировка имеет цис-конфигурацию.
Названия жиров составляются также как названия сложных эфиров, которыми они собственно и являются. При необходимости проставляются номера атомов углерода глицерина, при которых находятся соответствующие остатки высших жирных кислот. Так, жиры, формулы которых приведены выше, имеют следующие названия: тристеарат глицерина и 1-олеат-2-линолеат-3-линоленоат глицерина.
Химические свойства
Химические свойства жиров определяются сложноэфирным строением молекул триглицеридов и строением и свойствами углеводородных радикалов жирных кислот, остатки которых входят в состав жира.
Как сложные эфиры жиры вступают, например, в следующие реакции:
– Гидролиз в присутствии кислот (кислотный гидролиз)
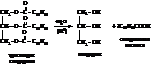
Гидролиз жиров может протекать и биохимическим путем под действием фермента пищеварительного тракта липазы.
Гидролиз жиров может медленно протекать при длительном хранении жиров в открытой упаковке или термической обработке жиров в условиях доступа паров воды из воздуха. Характеристикой накопления в жире свободных кислот, придающих жиру горечь и даже токсичность является «кислотное число»: число мг КОН, пошедшее на титрование кислот в 1г жира.
– Омыление:
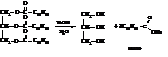
Мылами называют соли щелочных металлов жирных кислот, содержащих 10…18 углеродных атомов. Они имеют длинную, препятствующую растворению в воде углеводородную цепь, связанную со способствующим растворению карбоксилатным ионом, и поэтому действуют как смачивающие, эмульгирующие агенты и детергенты (моющие средства). Натриевые и калиевые мыла растворимы в воде и хорошо «мылятся». Калиевые соли высших жирных кислот дают жидкое мыло, натриевые - твердое. Соли магния, кальция, бария и некоторых других металлов очень плохо растворяются в воде; поэтому обычные мыла в жесткой воде переходят в нерастворимое состояние, не «мылятся», не пенятся, становятся липкими.
Наиболее интересными и полезными реакциями углеводородных радикалов являются реакции по двойным связям:
– Присоединение брома
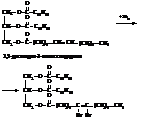
Степень ненасыщенности жира (важная технологическая характеристика) контролируется по «йодному числу»: число мг йода, пошедшее на титрование 100 г жира в процентах (анализ с бисульфитом натрия).
– Гидрогенизация жиров
Жидкие растительные масла (подсолнечное, хлопковое, соевое и другие) в присутствии катализаторов (например, губчатый никель) при температуре 175…190 °С и давлении 1,5…3,0 атм гидрируются по двойным С = С связям углеводородных радикалов кислот и превращаются в твёрдый жир – саломас. При добавлении к нему так называемых отдушек для придания соответствующего запаха и яиц, молока, витаминов и других ингредиентов для улучшения питательных качеств получают маргарин. Саломас используется также в мыловарении, фармации (основы для мазей), косметике, для изготовления технических смазок и т.д.
Пример реакции гидрогенизации:

– Окисление
Окисление перманганатом калия в водном растворе приводит к образованию насыщенных остатков дигидроксикислот (реакция Вагнера)

– Окислительное прогоркание жиров
Под действием влаги, света, повышенной температуры, а также следов железа, кобальта, меди, марганца в виде солей, содержащиеся в глицеридах остатки высших жирных кислот (прежде всего, ненасыщенных) медленно окисляются кислородом воздуха. Этот процесс протекает по цепному радикальному механизму и самоускоряется образующимися продуктами окисления. На первой стадии окисления кислород присоединяется по месту двойных связей, образуя пероксиды:
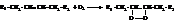 Кислород также может взаимодействовать с активированной
Кислород также может взаимодействовать с активированной
a-ме-тиленовой группой при двойной связи с образованием гидропероксидов:
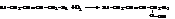
Пероксиды и гидропероксиды как соединения нестойкие разлагаются с образованием низкомолекулярных летучих кислород-содержащих соединений (спиртов, альдегидов и кетонов, кислот с углеродной цепочкой меньшей длины, чем в исходном жире, а также их разнообразных производных). В результате жир приобретает неприятный, «прогорклый» запах и вкус и становится непригодным для пищи.
Твердые, насыщенные жиры более устойчивы к прогорканию, хотя и в них могут образовываться гидропероксиды на базе a-углеродов в остатках кислот при сложноэфирной группировке жира. Для предотвращения окислительного прогоркания к жирам добавляют антиоксиданты.
При неправильном хранении жиры могут гидролизоваться с образованием свободных кислот и глицерина, что также изменяет их вкус и запах.
Хранить жиры необходимо в небольших темных склянках, доверху заполненных маслом, в сухом, прохладном, затемненном месте и в герметичной светонепроницаемой упаковке.
– «Высыхание» масел
Так называемые высыхающие масла состоят из глицеридов сильно ненасыщенных кислот (линолевой, линоленовой и др.) На свету под действием кислорода воздуха они окисляются и полимеризуются на поверхности в виде твёрдой эластичной плёнки. Процесс «высыхания» ускоряется катализаторами – сиккативами. Льняное масло, сваренное с оксидом или нафтенатами свинца (сиккатив) известно под названием олифа. Она применяется для приготовления масляных красок, линолеума, клеёнки и т.д.
АМИНЫ
Aминами называют, продукты замещения атомов водорода в аммиаке на углеводородные радикалы.
7.1 Алифатические амины
В противоположность, например, спиртам, галогеналканам, первичными аминами называют продукты замещения одного атома водорода в аммиаке R–NH2. Причём первичность, вторичность или третичность углеводородного радикала в данном случае роли не играет. Замещение двух водородов аммиака на одинаковые или разные углеводородные радикалы дает вторичный, а трех – третичный амин. Изомерия аминов определяется положением аминогруппы в углеродной цепи и количеством и строением радикалов, связанных с азотом.
По радикально-функциональной номенклатуре амины обычно называют как производные аммиака, добавляя к названию радикалов слово «амин». При составлении названия по номенклатуре ИЮПАК исходят из названия углеводорода с самой длинной цепью и вводят названия замещающей группы в этой цепи в виде приставки: амино-
(-NH2), метиламино- (-NHCH3), диметиламино- [-N(CH3)2] и т.д. Цифрой указывают положение заместителя.
Вот названия некоторых изомерных аминов С4Н9NH2:

| бутиламин, 1-аминобутан |
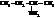
| втор-бутиламин, 2-аминобутан |
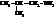
| изобутиламин, 1-амино-2-метилпропан |

| метилпропиламин, 1-метиламинопропан |

| диэтиламин, этиламиноэтан |

| метилизопропиламин, 2-метиламинопропан |

| диметилэтиламин, диметиламиноэтан |
Способы получения
– Главный путь получения аминов – алкилирование аммиака или аминов галоидными алкилами – реакция Гофмана.
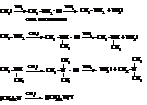
Таким образом, в зависимости от соотношения реагентов и условий реакции Гофмана получается смесь первичного, вторичного, третичного аминов и четвертичной соли аммония.
– Амиды кислот при расщеплении гипобромидом или гипохлоридом дают первичные амины
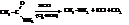
– Восстановление нитросоединений, нитрилов и изонитрилов.
Реакции протекают при действии водорода в присутствии катализаторов Pt, Pd, Ni.
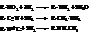
Физические свойства
Простейшие амины – газы, хорошо растворимые в воде и обладающие сходным с аммиаком запахом, но с более сильным «рыбным» оттенком. По сравнению со спиртами амины менее диссоциированы. Поэтому температуры кипения их соответственно ниже. Среди изомеров ниже кипят третичные амины, выше – первичные.
Химические свойства
В химическом отношении амины сходны с аммиаком.
– Также как и аммиак они обнаруживают щелочную реакцию с обычными индикаторами. Свойства аминов как оснований обусловлены способностью атома азота за счёт неподелённой пары электронов присоединять протон с образованием алкилзамещённого иона аммония
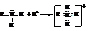
С водой амины образуют алкилзамещённые гидроксиды аммония
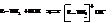
С минеральными кислотами они дают алкилзамещённые аммонийные соли
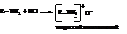
Реакции с кислотами свидетельствуют о том, что амины являются органическими основаниями. В большинстве случаев алифатические амины более сильные основания, чем аммиак. Являясь донорами электронов, алкильные радикалы повышают электронную плотность на азоте, усиливая при этом его способность захватывать и удерживать протон, что и демонстрирует свойства аминов как оснований. Так, константы основности K (NH3). = 1,79·10-5, K (CH3→NH2 ) = 4,38·10-4 и K (CH3→NH← CH3) = 5,2·10-4.
– Амины можно алкилировать (см. реакцию Гофмана).
– Амины подвергаются ацилированию, в том числе и ацетилированию уксусным ангидридом или хлористым ацетилом:
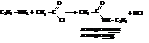
– Реакции с азотистой кислотой. Первичные, вторичные и третичные амины дают разные продукты.
При обработке первичных аминов азотистой кислотой выделяется свободный азот, и образуются спирты
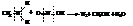
Вторичные амины дают нитрозоамины
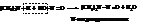
Третичные амины устойчивы к действию азотистой кислоты и образуют с ней соли.
– Соли четырёхзамещённого аммония. При нагревании третичных аминов с галоидными алкилами образуются соли четырёхзамещённого аммония
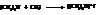
При действии на четвертичные соли аммония гидроксида серебра образуются четвертичные аммонийные основания – такие же сильные основания как едкий натр или едкое кали.
Амины нашли применение как органические основания и растворители, в синтезах некоторых поверхностно активных веществ, добавок при флотационном разделении солей, при получении целого ряда других ценных продуктов.
Диамины
Две первичные аминогруппы лишь в редких случаях удерживаются у одного атома углерода. Простейший первичный диамин, этилендиамин или 1,2-диаминоэтан, получают аммонолизом 1,2-дихлорэтана
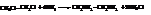
Примеры названий:
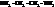
| этилендиамин, 1,2-диаминоэтан; |

| триметилендиами, 1,3-диаминопропан |

| пропилендиамин, 1,2-диаминопропан |

| гексаметилендиамин, 1,6-диаминогексан; |
Химические свойства диаминов повторяют свойства моноаминов; реагировать могут уже две аминогруппы.
В химической промышленности большое значение имеет гексаметилендиамин. Его получают из адипиновой кислоты.
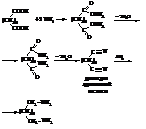
Поликонденсацией адипиновой кислоты и гексаметилендиамина получают высокомолекулярный полиамид с довольно высокой температурой плавления. Из расплава этого полимера формуют синтетическое волокно найлон, весьма близкое по своим свойствам к натуральному шёлку.
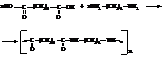
7.2 Ароматические амины
Ароматические амины, также как и амины жирного ряда, являются производными аммиака. Аминогруппа может быть соединена непосредственно с ароматическим ядром.
Амины с аминогруппой в боковой цепи, например бензиламин, похожи по своим свойствам на алифатические амины.
Ароматические амины могут быть первичными, вторичными и третичными
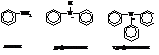
N-Метиланилин относится к жирноароматическим аминам
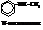
Названия аминов даются по тривиальной и ИЮПАК номенклатурам:

| Анилин, аминобензол |

| о-Толуидин или 1,2-толуидин, 2-метиланилин |
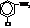
| м-Хлоранилин, 3-хлоранилин |

| о-Фенилендиамин, 1,2-диаминобензол |

| о- или 1,2-Аминобензолсульфокислота, ортаниловая кислота; м- или 1,3-Аминобензолсульфокислота, метаниловая кислота; п- или 1,4-Аминобензолсульфокислота, сульфаниловая кислота. |

| N,N-Диметиланилин |
Способы получения
Первичные амины
– Восстановление нитросоединений. В 1842 г. в лаборатории Казанского университета Н.Н. Зинин открыл метод получения анилина реакцией восстановления нитробензола сероводородом.

Только после этого открытия анилин приобрёл огромное промышленное значение. Он является исходным продуктом для получения многочисленных красителей, фармацевтических и взрывчатых веществ, фотореагентов.
По мере увеличения производства анилина на первый план выдвинулись каталитические и электрохимические методы восстановления.
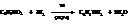
– Аминирование галогенпроизводных. Эта реакция в практическом отношении интересна вследствие дешевизны исходных продуктов: аммиака и галогенидов. Процесс проводится в автоклавах под давлением и при нагревании в присутствии медных катализаторов

Вторичные амины
– Смесь солей вторичных и третичных жирно-ароматических аминов в промышленности получают алкилированием первичных ароматических аминов галогеналканами или спиртами:
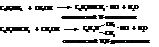 – Чисто ароматические вторичные амины получают нагреванием аминов с их солями
– Чисто ароматические вторичные амины получают нагреванием аминов с их солями
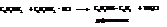
– Третичные амины, как чисто ароматические, так и жирно-ароматические, получают алкилированием первичных или вторичных аминов.

Физические свойства
Ароматические амины – это жидкие и твёрдые вещества, плохо растворимые в воде. Растворимость увеличивается при увеличении числа аминогрупп в ядре:
| tкип.( °С) | tпл.( °С) | |
| анилин | 184,4 | –6,2 |
| м-толуидин | 203,3 | –31,5 |
Химические свойства
– Анилин – основание гораздо более слабое, чем аммиак и жирные амины. Так, константа основности для метиламина 4,4·10-5, а для анилина – 3,8·10-10. Анилин слабо растворим в воде, его растворы не окрашивают лакмус и фенолфталеин. Он не образует солей со слабыми кислотами, такими как угольная и синильная. Ослабление основных свойств трёхвалентного азота в анилине вызвано смещением его неподелённой пары электронов к ядру и сопряжение её с π-электронами ядра. По этой причине электроны азота в меньшей степени способны к взаимодействию с протоном, то есть к реакции, характерной для оснований. Смещение электронов к ядру вызывает, кроме того, повышение электронной плотности в орто- и пара-положениях, что облегчает атаку электрофильных реагентов в эти положения. Присутствие электроноакцепторных групп в ядре уменьшает основность. Константа основности о-нитроанилина 10-14. В то же время, введение алкильных групп в аминогруппу увеличивает основнось. Kосн. N-метиланилина 7,1·10-10.
– С сильными кислотами амины образуют твёрдые соли, сильно гидролизованные в растворе
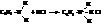
– Подобно аминам жирного ряда первичные и вторичные амины способны замещать атомы водорода аминогруппы на алкилы. Эта реакция уже упоминалась как метод синтеза вторичных и третичных аминов.
– Взаимодействие аминов с карбоновыми кислотами, их ангидридами и хлорангидридами приводит к замещению атома водорода в аминогруппе кислотными остатками. Это – реакция ацилирования.
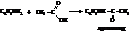
Ацильные производные аминов уже не обладают основными свойствами. Они устойчивы к окислителям. Поэтому ацильные остатки часто вводятся в амины временно для предохранения аминогруппы от окисления.
– Особенно большое значение в ароматическом ряду имеет реакции аминов с азотистой кислотой (азотистая кислота образуется при взаимодействии растворов нитрита натрия или калия с минеральными кислотами).
При действии азотистой кислоты на соли первичных аминов получаются соли диазония.
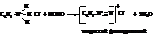
Жирные амины в этих условиях дают спирты (отличие !!!).
Вторичные ароматические амины с азотистой кислотой образуют N-нитрозосоединения, которые под действием минеральных кислот изомеризуются с перемещением нитрозогруппы в пара-положение ядра.
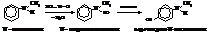
Третичные ароматические амины дают с азотистой кислотой пара-нитрозосоединения.

– Поскольку водородные атомы ядра в о- и п-положениях обладают в аминах большой подвижностью, амины легко вступают в различные реакции замещения.
– Галогенирование и нитрование анилина обычно даёт смесь продуктов различной степени замещения и сопровождается окислением. Поэтому для защиты аминогруппы галогенированию и нитрованию чаще всего подвергают их ацильные производные
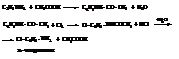
– При сульфировании получается также пара-изомер.
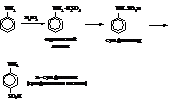
Основной областью использования анилина и многих его производных является анилино-красочная промышленность.
| <== предыдущая страница | | | следующая страница ==> |
| ЧАСТЬ 1 | | | АРОМАТИЧЕСКИЕ ДИАЗО- И АЗОСОЕДИНЕНИЯ, КРАСИТЕЛИ |
Дата добавления: 2014-10-02; просмотров: 1274; Нарушение авторских прав

Мы поможем в написании ваших работ!